Tema 1
- La solución al ejercicio 1.2 no debe considerarse la b, sino la d. Además, es preferible sustituir la explicación de la resolución del ejercicio que se da por esta otra:
«d. En general, cuando la fuente de contaminación está muy localizada en un punto y se puede extender en todas direcciones, el muestreo recomendable por su mejor relación resultados/coste es el polar. El inconveniente principal (que es que no todos los puntos dibujados sistemáticamente o seleccionados aleatoriamente en un plano de una ciudad son fácilmente muestreables, ya que algunos pueden coincidir con tejados o zonas particulares) no impide hacer un muestreo de este tipo, pues los puntos de muestreo no tienen por qué ser exactamente los inicialmente elegidos (no se espera un error apreciable por muestrear unos metros más allá del punto marcado en el plano). Un muestreo aleatorio nunca se puede decir que no dé buenos resultados, pero en este caso no es recomendable por la dificultad anterior y por su costo (se necesita tomar muchas muestras). El muestreo a juicio sería plausible si la ciudad estuviera sometida continuamente a vientos de una determinada componente, de modo que pudiéramos suponer que el contaminante solo se transporta hacia una dirección determinada».
Tema 2
- En la página 111, al final, se dice: «…si c0 en el patrón original es 0.1 M, las concentraciones de analito procedente del patrón en cada uno los recipientes (de izquierda a derecha en la figura 2.11) serán 0.01, 0.02, 0.03, 0.04 y 0.05 M». Lo correcto es: 0.00, 0.01, 0.02, 0.03 y 0.04 M.
- En las páginas 123-124 el ejercicio 2.6 debe renumerarse como 2.7 y viceversa.
Tema 3
- En el segundo párrafo de la página 147, donde dice «figura 3.21-derecha» debe decir «figura 3.22-derecha».
- Página 155. Al inicio del apartado 3.6.2, donde dice:
Sea P0 la potencia de la radiación de determinada l que sale de una fuente e incide en una muestra, y P la que sale de la muestra y alcanza el detector, de la misma l. El cociente P/P0 es la fracción de potencia total absorbida y se llama transmitancia (T), debe decir: «Sea P0 la potencia de la radiación de determinada l que sale de una fuente e incide en una muestra, y P la que sale de la muestra y alcanza el detector, de la misma l. El cociente P/P0 es la fracción de potencia total transmitida y se llama transmitancia (T)». - En esa misma página, arriba, al final del párrafo introductorio del apartado 3.6, donde dice:
«en absorción, la relación lineal lo es entre la concentración y el logaritmo de la fracción de potencia radiante absorbida por la muestra», debe decir: «en absorción, la relación lineal lo es entre la concentración y el logaritmo de la fracción de potencia radiante transmitida por la muestra«. - En la página 159, al principio del apartado 3.6.3 se dice: Aunque las expresiones [3.4] y [3.6] predicen teóricamente relaciones lineales entre la potencia absorbida o la absorbancia y la concentración…
pero debe decir: «Aunque las expresiones [3.4] y [3.6] predicen teóricamente relaciones lineales entre la potencia emitida o la absorbancia y la concentración…». - En la resolución del ejercicio 3.7 se ha empleado el valor de temperatura de 10000 K en vez del de 8150 que aparece en el enunciado.
Tema 4
- Página 189. En el penúltimo renglón se lee «radiación de sincrotón». Es «sincrotrón«.
Tema 5
- En la página 235 en la segunda línea, pone «figura 5.9». Lo correcto es: «figura 5.10».
Tema 7
- En la página 344, en su penúltima línea, dice «10, 4, 4, 4 y 3». Lo correcto es: «10, 4, 4, 4 y 6».
Tema 8
- En la página 372, en la línea 14ª, dice «dióxido de tritio», pero debe decir «óxido de tritio».
Tema 9
- En la página 407, en la figura, en la reacción del Zn, en la parte de la derecha de la flecha, el 2 que aparece como subíndice del Zn debe aparecer como superíndice.
- En la página 414, inmediatamente antes del epígrafe 9.3.1.2 dice: «De las reacciones que aparecen en la tabla se dan más fácilmente que la reducción del hidrógeno todas las reacciones de reducción que figuran por encima de la de él; es decir, todas las que tienen un valor E0 < 0″. Pero en realidad debe decir: «De las reacciones que aparecen en la tabla se dan más fácilmente que la reducción del hidrógeno todas las reacciones de reducción que figuran por debajo de la de él; es decir, todas las que tienen un valor E0 > 0″.
- Página 421. En el antepenúltimo párrafo se lee: «El electrodo selectivo de protones se conoce por el nombre de electrodo de vidrio porque se basa en una membrana de un material vítreo. Merece una atención especial». Aunque no se trata de una errata, la frase estaría mejor escrita así: «Se emplea para ello el llamado electrodo de vidrio porque se basa en una membrana de un material vítreo. (Existen electrodos de vidrio para determinar otros cationes, como Na+ o Ag+)».
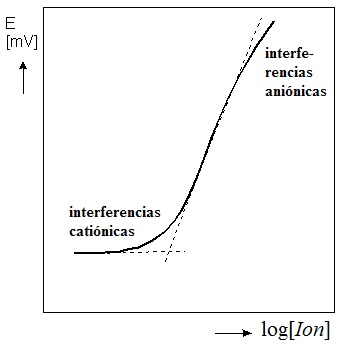
- Página 425. En el eje de abscisas de la gráfica 9.12 debe decir -pIon, no pIon o, lo que sería aun más claro: log[Ion]. Lo mismo en el pie de la figura.
- Página 433. La pregunta 9.5 («Uno de los siguientes dispositivos no es adecuado para medir directamente fluoruros en agua.») debe sustituirse por esta otra: «Solo uno de los siguientes dispositivos es adecuado para medir directamente cloruros en agua». La respuesta correcta (página 581) sería la b y debería estar acompañada por este texto: «b. El electrodo de vidrio mide la concentración de protones y otros cationes. Los electrodos señalados en a y c son específicos para el fluoruro».
Tema 10
- En el penúltimo párrafo de la página 464, donde dice «Esto es así porque Λ depende de la velocidad de migración de los iones y esta es mayor a menor dilución porque los iones encuentran menos impedimento en su recorrido» debe decir «Esto es así porque Λ depende de la velocidad de migración de los iones y esta es mayor a mayor dilución porque los iones encuentran menos impedimento en su recorrido».
- En el tercer párrafo de la página 467, donde dice «Basta medir la conductividad G«, debe decir «Basta medir la conductancia G«. En ese mismo párrafo, donde dice «conductancia equivalente» debe decir «conductividad equivalente».
- En la página 470, en el renglón 12, donde dice polarimetría debe decir polarografía.
Tema 11
- En la página 498, casi al comienzo, donde dice «…la representación de la masa frente al tiempo constituye un termogravigrama» debe decir más correctamente «…la representación de la masa frente a la temperatura constituye un termogravigrama» (como se puede observar en la figura que aparece más abajo).
- En la página 501, en la 2ª línea tras la figura, dice «todos exotérmicos, todos exotérmicos o de uno y…». Lo que debe decir es: «todos exotérmicos, todos endotérmicos o de uno y…».
Tema 12
 Página 423. La ecuación 9.18 no es pH = 0,059(K – Epila(medido)), sino pH = (K – Epila(medido))/0,059.
Página 423. La ecuación 9.18 no es pH = 0,059(K – Epila(medido)), sino pH = (K – Epila(medido))/0,059.- Página 425. En el eje de abscisas de la gráfica 9.12 debe decir -pIon, no pIon o, lo que sería aun más claro: log[Ion]. Lo mismo en el pie de la figura. (Ver imagen a la derecha).
- Página 433. La respuesta b a la pregunta 9.5 («Electrodo de segunda clase de plata») debe sustituirse por esta otra: «Un electrodo selectivo de iones especialmente diseñado».
- En la página 534, en el segundo renglón, donde dice «cada muestra» sería más propio decir «cada analito».
- En la página 545 (punto 12.4.4) se dice que «…se pueden distinguir varios tipos generales de cromatografía líquida en columna y en capa fina: de reparto, de adsorción, de intercambio iónico y de exclusión por tamaño«. Hay que aclarar que esas son variedades solo de la cromatografía en columna, no de la capa fina.
- Página 546. En el penúltimo párrafo, donde dice «un disolvente orgánico polar con metanol» debe decir «un disolvente orgánico polar, como el metanol,».
- En la página 550, en el quinto renglón, donde dice «lápiz de plomo» debe decir «lápiz de grafito».
- Página 557. En el antepenúltimo párrafo, donde dice «separar y cuantificar los aniones y cationes más comunes (halogenuros, NO3–, NO2–, SO42- o PO43…» falta la carga negativa del grupo fosfato (PO43-).

Glosario
- Página 592. En la definición de isótopo, donde se dice «se conocen dos isótopos estables del cloro: 35Cl y 37Cl. El primero tiene 35 neutrones y el segundo 37″ debería decirse «El primero tiene 18 neutrones y el segundo 20».



