




 En este enlace se puede encontrar:
En este enlace se puede encontrar:
- El prefacio del libro Técnicas Fisicoquímicas en Medio Ambiente
- El índice
- Las dos primeras páginas de cada tema
- La bibliografía
Comentarios desactivados en Extractos del libro
 Esta es la portada* de la edición de 2011 del libro Técnicas Fisicoquímicas en Medio Ambiente, que corrige las erratas y mejora algunos aspectos de la de 2007 (aunque en esencia pueden considerarse ambos libros la misma obra).
Esta es la portada* de la edición de 2011 del libro Técnicas Fisicoquímicas en Medio Ambiente, que corrige las erratas y mejora algunos aspectos de la de 2007 (aunque en esencia pueden considerarse ambos libros la misma obra).
La edición de 2007, de la que quedan pocos ejemplares en las librerías, recibió el premio a la mejor unidad didáctica de nueva edición en la convocatoria 2008 de estos galardones que entrega anualmente el Consejo Social de la UNED.
Aquí puede encontrarse un extracto de la edición de 2007 (muy parecida a la de 2011).
————————
Referencia bibliográfica:
Jose Mª Gavira Vallejo, Antonio Hernanz Gismero: Técnicas Fisicoquímicas en Medio Ambiente, Editorial UNED, Colección Grado, 6101206GR01A01, Primera edición: noviembre de 2011, ISBN: 978-84-362-6389-3
————————
*Foto de portada: Inmaculada García López (zeropuntonueve.blogspot.com)
————————
Librerías:
Comentarios desactivados en TFQMA – edición de 2011 (Grado)
 Esta es la portada de la edición de 2007 del libro Técnicas Fisicoquímicas en Medio Ambiente, que recibió el premio a la mejor unidad didáctica de nueva edición en la convocatoria 2008 de estos galardones que entrega el Consejo Social de la UNED.
Esta es la portada de la edición de 2007 del libro Técnicas Fisicoquímicas en Medio Ambiente, que recibió el premio a la mejor unidad didáctica de nueva edición en la convocatoria 2008 de estos galardones que entrega el Consejo Social de la UNED.
Agotada la edición se publicó otra en 2011 que prácticamente no difiere de la de 2007, siendo la novedad más destacada que se corregieron las erratas que se detectaron.
Aquí puede encontrarse un extracto de esta edición.
————————
Referencia bibliográfica:
José Mª Gavira Vallejo, Antonio Hernanz Gismero: Técnicas Fisicoquímicas en Medio Ambiente, Ed. UNED, 2007 (ISBN: 9788436255096), 616 páginas
Librerías:
UNED
Agapea.com
Amazon
Casa del Libro
Librería Gradua2
Librería Jurídica Tirant Lo Blanch
Libros Aula Magna
Prometeo Libros
Sanz y Torres
Comentarios desactivados en TFQMA – edición de 2007 (Licenciatura)
Tema 2
Variante para explicar la distribución log-normal
Tema 3
Un espectroscopio atómico casero
Otro. Se muestra su fabricación mediante un vídeo
Tema 4
Experimento de difracción de la luz de un puntero láser
Tema 5
Tema 6
Base de datos espectroscópicos
Otra base de datos espectroscópicos
Tema 7
——————————-
Por favor, si detecta algún enlace roto comuníquelo a tfqma@lajarda.com.
Comentarios desactivados en Webs para realizar las actividades
Por favor, si detecta algún enlace roto comuníquelo a tfqma@lajarda.com.
——————————-
Tema 1 (RECOGIDA Y TRATAMIENTO DE MUESTRAS AMBIENTALES PARA SU ANÁLISIS)
Tema 2 (QUIMIOMETRÍA)
Tema 3 (ESPECTROSCOPÍA ATÓMICA)
Tema 4 (FLUORESCENCIA, ABSORCIÓN Y DIFRACCIÓN DE RAYOS X)
Tema 5 (ESPECTROSCOPÍA DE ABSORCIÓN UV-VISIBLE Y DE LUMINISCENCIA)
Tema 6 (ESPECTROSCOPÍAS INFRARROJA Y RAMAN)
Tema 7 (RESONANCIA MAGNÉTICA NUCLEAR)
Tema 8 (ESPECTROMETRÍA DE MASAS)
Tema 9 (POTENCIOMETRÍA)
Tema 10 (TÉCNICAS DE CORRIENTE ELÉCTRICA)
Tema 11 (TÉCNICAS RADIOQUÍMICAS Y TÉRMICAS Y MÉTODOS AUTOMÁTICOS)
Tema 12 (TÉCNICAS CROMATOGRÁFICAS)
Tema 1 (RECOGIDA Y TRATAMIENTO DE MUESTRAS AMBIENTALES PARA SU ANÁLISIS)
- Página de enlaces sobre muestreo y análisis ambiental
- Guía de diseño de un muestreo ambiental
- Una introducción a los métodos de muestreo (en general, no solo en medio ambiente)
- Protocolos y métodos seguidos en España para analizar muestras ambientales
- Técnicas de muestreo (Instituto Nacional de Ecología, México)
- Fundamentos de diseño de muestreo
- Página de la Agencia de Protección Ambiental (EPA) de Estados Unidos
- Métodos de análisis de agua de la EPA
- Otros métodos de la EPA
- Programa de monitorización ambiental de la EPA
- Web de la Inspección Geológica de los Estados Unidos (USGS)
- Programa de calidad del agua de la USGS, con ejemplos de muestreo
- Equipos para muestrear agua (USGS)
- Lista muy completa de métodos de análisis ambiental de la NEMI
- Agencia Europea del Medio Ambiente (EEA)
- Página comercial de equipos de muestreo y análisis
- Trabajo sobre contaminación de un suelo por arsénico
- Técnicas de extracción de muestras de petróleo
^^^Volver al índice
Tema 2 (QUIMIOMETRÍA)
Sección de Estadística Ambiental de la ONU. Contiene tablas de indicadores ambientales de todo el mundo, informes (también en español) y un interesante glosario ambiental (unstats.un.org/unsd/environmentgl/).
Buena página sobre distintos métodos estadísticos.
Comparación de las distribuciones de Student y gaussiana y tablas de valores de t.
Centro de Ecología Estadística y Estadística Ambiental de la Universidad de Pensilvania. De él se pueden descargar muchos informes técnicos de interés (www.stat.psu.edu/~gpp/technical_reports.htmy www.stat.psu.edu/~gpp/encyclopedia_entry.htm)
Un interesante artículo que explica por qué muchas variables siguen un comportamiento log-normal (Limpert, 2001).
Varios sitios que contienen estadísticas de calidad ambiental.
Gráficos de control ambiental.
Calculadoras en línea
- www.math.csusb.edu/faculty/stanton/m262/regress/regress.html
- www.stat.wvu.edu/SRS/Modules/Applets/Regression/regression.html
Applets para realizar la regresión lineal de una serie de puntos que el usuario va marcando en pantalla mediante el ratón o escribiendo en una ventana de datos; se obtiene la recta de regresión, su error y el coeficiente de correlación.
Más calculadoras estadísticas en línea.
Actividades
- webphysics.davidson.edu/applets/Galton/BallDrop.html
- www.inf.ethz.ch/personal/gut/lognormal/index.html
Tableros de Galton
^^^Volver al índice
Tema 3 (ESPECTROSCOPÍA ATÓMICA)
Animaciones para entender el espectro atómico del hidrógeno.
Excelente resumen de la espectroscopía atómica con esquemas y dibujos muy atractivos.
- www.shsu.edu/~chm_tgc/sounds/pushmovies/ICPwRC.gif
- www.shsu.edu/~chm_tgc/sounds/pushmovies/ICPwCCD.gif
Animaciones para entender la técnica ICP de detección simultánea de varios elementos y el uso de un detector de acoplamiento de carga.
Otra animación, en este caso para explicar un experimento de absorción atómica.
Interesante explicación de cómo se pone en práctica la técnica de generación de hidruros.
Espectros de emisión y absorción atómica de todos los elementos de la tabla periódica (se obtienen simplemente “pinchando” el elemento en la tabla).
Se pueden visualizar los colores que un buen número de elementos químicos dan a la llama.
Página que detalla pormenorizadamente las interferencias que puede sufrir cada elemento químico en espectroscopía atómica.
Informe completo del accidente de Aznalcóllar.
- www.montes.upm.es/Dptos/DptoIngForestal/OperacionesBasicas/Docencia/PDF/Presentaciones/Tema%2011.pdf
Un buen resumen sobre espectroscopía de absorción atómica.
Calculadoras en línea
Se trata de una calculadora de la distribución de Boltzmann a partir de la diferencia de energía entre dos estados y la temperatura, así como un convertidor de unidades de energía.
Actividades
Un espectroscopio atómico casero.
Otro. Se muestra su fabricación mediante un vídeo.
^^^Volver al índice
Tema 4 (FLUORESCENCIA, ABSORCIÓN Y DIFRACCIÓN DE RAYOS X)
- fr.wikipedia.org/wiki/Spectrom%C3%A9trie_de_fluorescence_X
- en.wikipedia.org/wiki/X-ray_fluorescence
- en.wikipedia.org/wiki/X-ray_crystallography
Excelentes compendios de la Wikipedia (en inglés y francés) sobre las principales técnicas espectroscópicas de rayos X.
Rayos X en general. Muy buenos resúmenes básicos de todas las técnicas, incluida la SEM/EDX.
Buena explicación de las técnicas de espectroscopía de absorción de rayos X de estructura fina.
“Animación” sobre la espectroscopía de absorción de rayos X.
Página del Departamento de Cristalografía del CSIC dedicada a la difracción de rayos X. Tiene una introducción general con algunas animaciones muy aclaratorias.
Animaciones que ilustran cómo se obtiene el espectro de difracción.
Una página dedicada a los aspectos prácticos de la difracción de rayos X al alcance de todos.
X-ray based methods of analysis, de K. Janssens. Todas las técnicas de rayos X, con aplicaciones en el campo de la arqueología
Una aplicación en medio ambiente de la fluorescencia de rayos X.
Todos los aspectos prácticos sobre la preparación de la muestra para un espectro de rayos X.
Espectros de fluorescencia de rayos X de todos los elementos químicos, seleccionándolos en una tabla periódica interactiva.
Difracción de rayos X… y algunas cosas más.
Una base de datos de difracción de rayos X y de espectros Raman (tema 6).
Actividades
Experimento de difracción de la luz de un puntero láser.
^^^Volver al índice
Tema 5 (ESPECTROSCOPÍA DE ABSORCIÓN UV-VISIBLE Y DE
LUMINISCENCIA)
Una prolija explicación de por qué las cosas tienen el color que vemos.
Excelente resumen de la técnica de espectroscopía UV-visible.
Explicación de las diferencias entre fluorescencia y fosforescencia.
Muy cuidada explicación con voz (inglés) y animaciones de la radiación electromagnética y la fluorescencia.
Resumen esquemático de la fluorimetría.
En estas páginas se pueden consultar los espectros de absorción UV-visible y fluorescencia molecular (emisión y excitación) de muchas especies de interés en bioquímica.
- www.shsu.edu/~chm_tgc/sounds/pushmovies/DB.gif
- www.shsu.edu/~chemistry/spectrometer/spect.gif
- www.wooster.edu/chemistry/is/brubaker/uv/uv_works_modern.html
Animaciones que ilustran el funcionamiento de un espectrómetro UV de doble haz y de matriz de diodos.
Esta página, excelente, reúne una serie de recursos sobre espectroscopía molecular. Aunque en este momento solo interesa el apartado de espectroscopía UV, conviene volver a esta dirección cuando se estudien las espectroscopías infrarroja y de resonancia magnética nuclear y la espectrometría de masas.
Buena información sobre todos los tipos de sensores.
Una clara introducción a la inmunoquímica, en español.
Aplicaciones de los biosensores inmunoquímicos en medio ambiente.
Se trata de un informe sobre los sensores químicos que se emplean para el análisis in situ de contaminantes volátiles. Conviene detenerse en los sensores ópticos.
Software
PhotochemCAD es un programa (gratuito) que permite relacionar la estructura de un buen número de compuestos orgánicos con su espectro UV-visible. Además, permite manipular estos espectros.
Es una hoja de cálculo MS Excel® que permite simular la forma de un espectro introduciendo los datos de la anchura y la intensidad de las bandas. Ayuda a entender el problema del solapamiento de las bandas espectrales.
Actividades
Por qué el agua de un vaso es incolora y sin embargo las grandes masas de agua se ven azules.
^^^Volver al índice
Tema 6 (ESPECTROSCOPÍAS INFRARROJA Y RAMAN)
Resumen muy claro de la espectroscopía IR con una animación que muestra los modos de vibración de la molécula de formaldehído y cómo se reflejan en su espectro.
Buen resumen de la técnica en español, con vibraciones en movimiento.
Interesante comparación, animada, de las espectroscopías IR y Raman.
- www.gps.caltech.edu/~edwin/molecular_vibrations.htm
- www.lsbu.ac.uk/water/vibrat.html
- www.umass.edu/microbio/chime/ir-spect/index.htm
- www.chem.umass.edu/~nermmw/Spectra/vibtutor.htm
- wwwchem.uwimona.edu.jm:1104/spectra/iranim/index.html
En estas páginas pueden encontrarse animaciones de vibraciones moleculares. En una de ellas, marcando en un espectro cada banda se puede observar qué movimiento vibratorio de la molécula da lugar a ella.
- www.chem.ucalgary.ca/courses/351/Carey/Ch13/ch13-ir-4.html
- www.colby.edu/chemistry/JCAMP/IRHelperNS.html
Ejemplos guiados de interpretación de espectros IR.
Se trata de un convertidor de energía / longitud de onda / frecuencia que facilita la comparación de las escalas IR y UV.
Es una base de datos excelente que contiene unos 50000 espectros infrarrojos y 3500 Raman. No solo es útil en este tema, sino en los siguientes, ya que asimismo recoge espectros de resonancia magnética nuclear (más de 27000) y de masas (más de 24000).
Ayuda para la interpretación de espectros IR.
Una forma muy sencilla y rápida de conocer los grupos químicos implicados en vibraciones para un número de ondas dado. (También da los protones para un desplazamiento químico RMN-1H que se introduzca y los fragmentos moleculares para valores m/q en espectrometría de masas.)
Software
Excelente programa multimedia sobre espectroscopía IR. Descargar en una carpeta todos los ficheros y ejecutar Irtutor.exe.
Lecturas
Artículo de Collette, T. W. y Williams, T. L.: The role of Raman spectroscopy in the analytical chemistry of potable water. J. Environ. Monit. 4 (2002) 27–34.
Actividades
Bases de datos espectroscópicos.
^^^Volver al índice
Tema 7 (RESONANCIA MAGNÉTICA NUCLEAR)
Muy completo. Se explican las bases matemáticas, físicas y químicas de la resonancia magnética nuclear, con sus aplicaciones. Se recurre para ello a claras animaciones y gráficos.
Excelente revisión de la técnica, con glosario, exposiciones gráficas y un vídeo.
Una página muy interesante desde el punto de vista aplicado, en especial el apartado dedicado a las diversas técnicas uni- y bidimensionales en RMN (chem.ch.huji.ac.il/nmr/techniques/expts.html).
Comparación de las técnicas de RMN, IR, UV y masas; lo que se puede obtener con cada una de ellas.
Una explicación muy completa de la técnica.
Animaciones para explicar el fundamento de la técnica.
Buen resumen, con gráficos muy claros.
- www.chemistry.vt.edu/chem-dept/hbell/simulation/hb2/ftsimstuff/simulateinfo.htm
- www.chemistry.vt.edu/chem-dept/hbell/simulation/hb2/vtftstuff/ftpackage.htm
Animaciones que explican paso a paso cómo se obtiene un espectro de RMN.
¡RMN y música! En esta página se puede oír la señal de decaimiento de la inducción (FID) de varias sustancias químicas.
Software
Se pueden encontrar programas de simulación de espectros RMN de la empresa española Mestrelab Research.
Actividades
Comprobación de cómo distintos sustituyentes afectan a los desplazamientos químicos de 13C de varios núcleos aromáticos (benceno, piridina, bifenilo…).
^^^Volver al índice
Tema 8 (ESPECTROMETRÍA DE MASAS)
Muy buena página en español de los fundamentos de la técnica y sus posibilidades.
Excelente resumen sobre espectrometría de masas, con interpretación de algunos espectros.
Buenos esquemas de métodos de fragmentación.
Una buena colección de espectros donde se ilustran los esquemas de fragmentación.
Una explicación de cómo funciona el espectrómetro de masas y de las características de los espectros.
Software
Puede encontrarse el programa Surface Spectra que permite predecir los posibles picos moleculares de un espectro de masas en función de todas las combinaciones de isótopos posibles que dan lugar a una determinada especie química. Por ejemplo, el Cl2 puede aparecer en la naturaleza en forma de 35Cl-35Cl, 35Cl-37Cl y 37Cl-37Cl, cuyos pesos moleculares respectivos aproximados son 70, 72 y 74; el programa calcula la probabilidad de existencia de esas tres especies.
^^^Volver al índice
Tema 9 (POTENCIOMETRÍA)
Una revisión en español de la potenciometría y sus aplicaciones analíticas
Es un buen esquema introductorio de electroquímica para entender sus bases.
Todo sobre los electrodos selectivos de iones.
Muy completo; todo sobre la potenciometría práctica.
Software
Puede encontrarse la hoja de cálculo MS Excel® Valpotenc que permite encontrar los puntos de equivalencia de una valoración potenciométrica mediante el método de las derivadas.
^^^Volver al índice
Tema 10 (TÉCNICAS DE CORRIENTE ELÉCTRICA)
- chemistry.about.com/library/weekly/aa082003a.htm
- www.sparknotes.com/chemistry/electrochemistry/electrolytic/section1.html
Se explica la diferencia entre una pila y una celda electrolítica.
Un repaso a los métodos voltamperométricos.
Cómo obtener información analítica de la voltamperometría.
Una excelente introducción a la voltamperometría cíclica.
Explicación del funcionamiento del biosensor amperométrico de determinación de glucosa.
Ejemplo de aplicación de los métodos electromagnéticos para estudios de suelos.
Ejemplos de valoraciones conductimétricas comparadas con las potenciométricas.
Artículos de aplicación de estas técnicas.
Software
Aquí puede encontrarse la hoja de cálculo MS Excel® Valconduc que permite averiguar el punto de equivalencia de una valoración conductimétrica.
Se puede encontrar una calculadora en línea que predice la conductividad de un agua natural según los sólidos totales disueltos que contenga.
Una calculadora para cambiar de unidades de conductividad.
^^^Volver al índice
Tema 11 (TÉCNICAS RADIOQUÍMICAS Y TÉRMICAS Y MÉTODOS
AUTOMÁTICOS)
Una versión bien explicada y documentada del accidente nuclear de Chernóbil.
Un buen resumen de las técnicas radioquímicas.
Espectros gamma de muchos isótopos.
Una página muy completa sobre los métodos térmicos de análisis.
Presentación esquemática del método de inyección de flujo.
Es una documentación excelente sobre aplicaciones ambientales de sistemas automáticos y determinación elemental por combustión. En general, toda la página www.uga.edu/~sisbl/#Erba ofrece contenidos muy interesantes en análisis ambiental.
Comparación de los métodos de flujo continuo y detenido para medidas cinéticas.
^^^Volver al índice
Tema 12 (TÉCNICAS CROMATOGRÁFICAS)
Biografía de Tsvet, descubridor de la cromatografía. Excelente artículo global.
Un buen resumen esquemático de las técnicas cromatográficas con dibujos y esquemas clarísimos (autora: Mª Paz Arraiza).
Un excelente fichero flash sobre aplicaciones de las cromatografías de gases y HPLC a estudios ambientales. (Se ha de descargar y ejecutar el fichero.)
Animaciones del funcionamiento de cromatógrafos (y de otros instrumentos).
Aplicaciones de la cromatografía de gases a la separación de hidrocarburos.
“Todo” sobre la cromatografía HPLC.
Una delicada aplicación: los perfumes.
- www.ugr.es/~quiored/lab/oper_bas/crom.htm
- latina.chem.cinvestav.mx/RLQ/tutoriales/cromatografia/Thin.htm#cuali
Buena página de cromatografía en capa fina en español.
Un buen artículo sobre las posibilidades de la electroforesis capilar en medio ambiente.
^^^Volver al índice
——————————-
Por favor, si detecta algún enlace roto comuníquelo a tfqma@lajarda.com.
Comentarios desactivados en Webs recomendadas para profundizar en los temas del libro
Tema 1 (RECOGIDA Y TRATAMIENTO DE MUESTRAS AMBIENTALES PARA SU ANÁLISIS)
Tema 2 (QUIMIOMETRÍA)
Tema 3 (ESPECTROSCOPÍA ATÓMICA)
Tema 4 (FLUORESCENCIA, ABSORCIÓN Y DIFRACCIÓN DE RAYOS X)
Tema 5 (ESPECTROSCOPÍA DE ABSORCIÓN UV-VISIBLE Y DE LUMINISCENCIA)
Tema 6 (ESPECTROSCOPÍAS INFRARROJA Y RAMAN)
Tema 7 (RESONANCIA MAGNÉTICA NUCLEAR)
Tema 8 (ESPECTROMETRÍA DE MASAS)
Tema 9 (POTENCIOMETRÍA)
Tema 10 (TÉCNICAS DE CORRIENTE ELÉCTRICA)
Tema 11 (TÉCNICAS RADIOQUÍMICAS Y TÉRMICAS Y MÉTODOS AUTOMÁTICOS)
Tema 12 (TÉCNICAS CROMATOGRÁFICAS)
Tema 1 (RECOGIDA Y TRATAMIENTO DE MUESTRAS AMBIENTALES PARA SU ANÁLISIS)
-
COMENTARIOS
Muestreo
Este libro trata de las técnicas y los instrumentos en los que se basa el análisis químico, particularmente de sustancias de interés en medio ambiente. En los temas 2 a 12 se explican las bases teórica de las técnicas fundamentales y cómo se aplican en los laboratorios. Hoy día la mayoría de estas técnicas han alcanzado tales niveles de perfección que el error que se comete al analizar una determinada muestra es, como mucho, del orden del 1%. Sin embargo, eso no garantiza que siempre vayamos a dar la concentración de un determinado analito en un sistema ambiental con tan escaso error. De hecho, el error puede ser mayor en varios órdenes. ¿Por qué? Un ejemplo lo aclarará.
Supongamos que queremos conocer la concentración media en determinado momento de cierto metal en el tramo de un río que se extiende desde el punto en que una factoría vierte sus residuos en el cauce hasta 1 kilómetro aguas abajo. No cometeríamos error si fuésemos capaces de trasvasar toda el agua contenida en ese tramo a un enorme contenedor, agitarla para lograr una homogeneización perfecta, tomar una alícuota y analizarla. Como, evidentemente, ese procedimiento es inviable por su coste, tenemos que ingeniárnoslas para obtener los mismos resultados a partir de un número limitado de muestras de agua del río. La ciencia de la estadística ha estudiado este problema y ha propuesto métodos para calcular el número de muestras necesarias y de dónde han de ser tomadas para que el conjunto de esas muestras representen al tramo del río en su totalidad.
Piénsese que se toman solo muestras junto a la boca de la cañería o bien que se toman 1 km aguas abajo. En ninguno de los casos los resultados coincidirán con la concentración media del contaminante en ese tramo. ¿Cómo hacer, entonces, para tener un alto nivel de confianza en que la media de concentración de las muestras que se toman es lo más parecida posible a la media real? Es decir, ¿cómo tomar las muestras de forma representativa?
El muestreo llamado a juicio o de conveniencia difícilmente va a garantizar la representatividad de la muestra, si bien tiene la ventaja de una estimación rápida de la situación. Frente a ese tipo de muestreo, existen otros más científicos como el aleatorio, el sistemático y el estratificado.
El muestreo aleatorio consiste en tomar las muestras en puntos del sistema determinados al azar. El inconveniente es que algunas áreas pueden no ser muestreadas. Para evitarlo se puede muestrear sistemáticamente, lo cual consiste en tomar muestras de cada sector dentro de una rejilla más o menos regular. En ocasiones, el sistema presenta claramente discontinuidades; es decir, hay zonas en que el analito está en una concentración que es claramente diferente a la que tiene en otra zona. Dicho de otro modo, pueden delimitarse regiones homogéneas dentro del sistema a las que se les llama estratos. Cuando se muestrea según las características de cada estrato (más muestras cuanto mayor es) el muestreo se llama estratificado. Existen otras variedades de muestreo híbridas de las anteriores.
Cada muestra que se toma de un sistema es una muestra simple. Si se analizan todas las muestras simples y se ha anotado en un mapa de dónde se tomó cada una, puede mapearse la presencia del analito en el sistema ambiental. Es decir, se puede conocer su distribución espacial (si se muestrea en el tiempo, su distribución temporal). Otra opción es analizar una sola muestra compuesta obtenida al mezclar previamente todas las muestras simples de modo que se obtenga un conjunto perfectamente homogéneo. Procediendo de este modo se puede obtener una media “física” de la propiedad del analito que se esté midiendo (por ejemplo, su concentración). Si las cosas se hacen bien, la media matemática de la propiedad del analito en todas las muestras simples y la media fisica obtenida tras formar previamente una mezcla compuesta no deben diferir excesivamente.
Instrumental y procedimientos de toma de muestra y transporte al laboratorio
Existen normas sobre el instrumental adecuado para recoger las muestras que se deben seguir estrictamente sobre todo cuando se quieren analizar trazas. Por ejemplo, no conviene emplear una pala niquelada para investigar la presencia de trazas de níquel en un suelo. Para analizar agua en busca de metales, los recipientes es conveniente que sean de plástico (excepto si se quiere analizar mercurio), pero si se han de determinar sustancias orgánicas, conviene recoger las muestras en recipientes cerámicos o de vidrio. Esto es así porque los plásticos liberan sustancias orgánicas y los vidrios, metales. Las precauciones han de ser más estrictas cuando se analizan trazas.
La muestra, si no se analiza in situ, ha de transportarse al laboratorio cuanto antes para evitar pérdidas (volatilización, absorción, adsorción, difusión…), degradaciones (oxidación, reacciones fotoquímicas, por microorganismos…) y contaminación del analito por otras sustancias.
Preparación de la muestra para el análisis
No se analiza habitualmente toda la muestra que se lleva a un laboratorio. Se suelen llevar cientos de gramos o kilos y normalmente con un gramo o un miligramo basta. Por ello, cada muestra hay que homogeneizarla perfectamente para que esa pequeña cantidad de ella que se toma sea verdaderamente representativa. Existen procedimientos normalizados para llevar a cabo esta tarea según la muestra sea sólida, líquida o gaseosa.
Por otro lado, normalmente, a la hora de analizar un analito suelen estorbar los demás presentes en la muestra, es decir, la matriz. Por ello, la muestra se suele someter a tratamientos para extraer el analito de su matriz. Cuando se trata de extraer sustancias orgánicas se emplean simples embudos de decantación o instrumental más sofisticado como columnas, jeringas de microextracción (si las muestras son líquidas) y soxhlets, baños de ultrasonidos, fluidos supercríticos, ec. (si son sólidas). Las muestras inorgánicas normalmente precisan ser puestas en disolución. Para ello se emplean ácidos, ultrasonidos o microondas o bien se funde la muestra, se mineraliza, se digiere…
Una vez extraído el analito, suele ser imprescindible concentrarlo. Para ello se pueden emplear aparatos idóneos como los rotavapores.
MATERIAL DIDÁCTICO
Ejemplos de aplicación de toma de muestras ambientales:
-
^^^Volver al índice
Tema 2 (QUIMIOMETRÍA)
- ErroresCuando se mide una variable en el medio ambiente (por ejemplo, la concentración de un contaminante) no basta dar el valor de la medida, sino que hay que acompañarlo del error que se estima que se ha cometido. Para hacer esta estimación no se siguen criterios subjetivos; al contrario, se hace uso de herramientas bien establecidas por la Estadística y de reglas seguras deducidas de la experiencia..Existen muchas causas por las que se comete error, pero en general cabe diferenciar las aleatorias de las sistemáticas. Los errores sistemáticos normalmente se pueden prever y corregir. Un error sistemático es, por ejemplo, el de una balanza descalibrada que pesa por exceso. La experiencia en el uso de esa balanza permite en muchos casos corregir el error.Sin embargo, los errores aleatorios, como su nombre indica, son imprevisibles y — precisamente por su aleatoriedad– cuando se hace la medida repetidas veces sobre una misma muestra se obtiene aproximadamente el mismo número de valores por encima que por debajo del valor verdadero o, en todo caso, el número de valores por encima supera ligeramente al de valores por debajo. Modelos muy comunes de distribuciones de datos del primer tipo (es decir, simétricas respecto al valor verdadero) son las de Gauss (también llamada normal) y de Student. Del segundo tipo (asimétricas), en medio ambiente la más común es la llamada log-normal.
No se suele hacer una sola medida instrumental para medir una variable de una muestra. O bien la medida se repite varias veces o bien se hace sobre varias alícuotas (partes de una muestra homogénea). De este modo, puede obtenerse una media. Ahora bien, no basta dar la media. Es necesario dar también un parámetro que informe de la magnitud de los errores aleatorios inherentes a la técnica o al método que se esté empleando. Este parámetro es la desviación típica. Si todas las medidas coinciden, su media, evidentemente, es ese mismo valor. La desviación típica, en ese caso, es cero. Por el contrario, cuanto más difieran los valores individuales, mayor será la desviación típica. (Este parámetro se llama así porque informa de lo que los datos individuales, en conjunto, se desvían o difieren de la media). Una media que lleva asociada una desviación típica alta no es fiable.
En química analítica se distingue entre exactitud de una medida (el grado de aproximación al valor verdadero; es decir, el error) y la precisión de una serie de medidas (la desviación típica). Un instrumento puede dar medidas precisas aunque no las dé exactas. Para conocer cuán exacto es un instrumento al medir, basta hacer la medida de un patrón. Para saber cuán preciso es, basta hacer varias medidas sobre la misma muestra o sobre alícuotas y calcular la desviación típica. Un método que proporciona medidas precisas en las mismas condiciones de operador, aparato, laboratorio, etc., se dice que tiene buena repetibilidad; cuando las da en condiciones distintas se dice que es reproducible.
Conocer aunque sea aproximadamente la desviación típica de una variable en un sistema ambiental (es decir, la variabilidad del sistema) es muy útil para calcular el número de muestras que habría que tomar para obtener una media significativa, es decir, para poder gozar de un cierto nivel de confianza de que la media obtenida es fiable.
Cuando se quiere monitorizar un sistema ambiental es muy útil llevar gráficos de control. En estos se va reflejando continuamente el valor medido de la variable de interés de modo que en todo momento se puede hacer una comparación visual de este valor con la media habitual y su desviación típica. De este modo puede constatarse fácilmente cuándo el sistema queda «fuera de control».
Calibración
El método general para determinar el valor de determinada variable en una muestra ambiental (por ejemplo, la concentración de cierto analito) es hacer previamente una serie de medidas de esa variable sobre otros tantos patrones y representar la señal analítica frente a tales valores. Uniendo los puntos se obtiene la curva de calibración (en realidad, más que gráficamente, se suele determinar mediante un procedimiento matemático llamado regresión). Después se mide la señal analítica que genera la muestra y este valor se interpola en la curva de calibración.
La curva de calibración ideal es la línea recta porque es la que produce menos errores a la hora de hacer la interpolación. Ahora bien, las curvas de calibración suelen dejar de ser rectas a altas concentraciones y a muy bajas (por distintas razones que dependen de cada técnica). Una técnica analítica es tanto más preferible cuanto mayor es el intervalo de concentraciones dentro del cual la señal analítica sigue un comportamiento lineal con la concentración. Hay técnicas que tienen un comportamiento lineal en un intervalo de siete órdenes de magnitud (por ejemplo desde 10^(-3) M a 10^3 M).
La pendiente de la recta de calibración da la sensibilidad del método empleado. Cuanto mayor es la pendiente, el error en la medida de la señal genera menos error en la medida de la concentración. No hay que confundir sensibilidad con límite de detección (aunque la mayoría de los libros incurren en ese error). El límite de detección de una técnica es la cantidad mínima de analito que la técnica detecta con un nivel de confianza dado. Son más adecuadas la técnicas cuanto más bajo límite de detección las caracterice y cuanta más alta sensibilidad tengan.
Una forma alternativa de construir la curva de calibración es, en vez de medir la señal en patrones, medirla en la propia muestra, a varias de cuyas alícuotas se agregan distintas cantidades de un patrón. Este método, llamado de adición de patrones, minimiza los efectos de matriz, es decir los errores inducidos por la presencia de otros analitos que interfieren en la determinación analítica del analito de interés. Una variedad de este método se conoce por el nombre de patrón interno.
^^^Volver al índice
Tema 3 (ESPECTROSCOPÍA ATÓMICA)
-
COMENTARIOS
Espectros atómicos
Tras los dos temas introductorios, a partir de este tercero se tratan propiamente las técnicas instrumentales de análisis químico, que aventajan a las técnicas clásicas (las marchas analíticas, las volumetrías, las gravimetrías) en multitud de aspectos. Así, las técnicas instrumentales son más rápidas, más automatizables, sus límites de detección son más bajos, se pueden determinar más analitos y los intervalos de comportamiento lineal de la señal con la concentración son más anchos.
Dentro de las técnicas instrumentales cabe diferenciar las espectroscópicas de las eléctricas y de otros tipos. Por otro lado, la cromatografía es una técnica especial que se apoya en las demás.
Las técnicas espectroscópicas se basan en la interacción de la radiación electromagnética con la materia. En general, irradiando una muestra se producen en ella cambios que permiten identificar las moléculas de que está formada e incluso su concentración.
La primera técnica de este tipo que se considera en este manual es la espectroscopía atómica. Se basa en la capacidad que tienen los átomos de absorber radiación electromagnética y de, posteriormente, emitirla. Lo interesante es que cada tipo de elemento químico absorbe y emite radiaciones de longitudes de onda características y diferentes a las longitudes de onda absorbidas o emitidas por otros elementos. Eso permite identificar el elemento. Además, cuanto más concentrado esté, más cantidad de radiación absorbe. Eso permite cuantificar el elemento.
Existen tres variedades de espectroscopía atómica: de absorción, de emisión y de fluorescencia. Las más aplicadas son las dos primeras. En espectroscopía atómica de absorción la muestra se atomiza calentándola en una llama o en un horno de grafito o sometiéndola a una descarga eléctrica. Después se irradia con radiaciones de diferentes longitudes de onda para observar cuáles de ellas son absorbidas (y en qué medida) y cuáles no. La representación gráfica de la cantidad de radiación absorbida (eje Y) de cada longitud de onda frente a dicha longitud de onda (eje X) es el espectro de absorción. Cada elemento químico tiene un espectro de absorción diferente. El espectro de absorción es como si fuese la huella dactilar de un elemento químico.
En espectroscopía atómica de emisión la muestra se atomiza y se excita (es decir, se deja que absorba radiaciones) normalmente en un plasma muy energético. Después se permite que se desexcite o relaje emitiendo las radiaciones que le son características. La medida de estas radiaciones (intensidad frente a longitud de onda) es el espectro de emisión.
Por su lado, en espectroscopía de fluorescencia la muestra se atomiza e irradia como en espectroscopía de absorción pero no se observa cuánto absorbe, sino cuánto emite posteriormente. Es un tipo de espectroscopía, pues, híbrido entre la de absorción y la de emisión.
Los espectros de absorción, emisión y fluorescencia normalmente están formados por una serie de picos. Los espectros se obtienen en aparatos llamados espectrómetros, que en general se componen de una fuente (para excitar la muestra), un monocromador (para seleccionar longitudes de onda individuales a partir de una radiación policromática) y un detector (para medir la intensidad de la radiación de cada longitud de onda que le llega).
Análisis cualitativo y cuantitativo
En absorción atómica, una medida de la cantidad de radiación absorbida por la muestra es la llamada absorbancia, que se define de tal modo que es linealmente proporcional a la concentración del analito (ley de Beer). En emisión y fluorescencia, la potencia de la radiación emitida a cada longitud de onda es también linealmente proporcional a la concentración.
Esto es en teoría. En la práctica, diversos inconvenientes hacen que estas relaciones no sean siempre lineales. Entre tales inconvenientes cabe destacar las interferencias, que son fenómenos diversos que afectan a la señal del analito aumentándola o disminuyéndola. En espectroscopía atómica son interferencias la superposición en el espectro de los picos de absorción o emisión de más de dos analitos, la aparición de un fondo continuo o bandas debidas normalmente a una atomización incompleta, la coexistencia en la llama o en el plasma de átomos de un elemento con iones del mismo, la formación de especies volátiles, la formación de especies refractarias o muy estables en general, etc. Se han ideado métodos para evitar todas las interferencias conocidas. En general, las interferencias pueden paliarse añadiendo especies químicas llamadas modificadores de matriz (supresores de ionización, agentes liberadores, agentes protectores…).
En medio ambiente la espectroscopía atómica es una técnica ideal para identificar metales tanto en suelos como en aguas y aire.
MATERIAL DIDÁCTICO
En los siguientes enlaces pueden encontrarse «notas de aplicación» de la espectroscopía atómica a problemas ambientales. (Consideramos que las «notas de aplicación» son un excelente recurso didáctico para aprender cómo se aplica en la práctica toda esta teoría.)
^^^Volver al índice
Tema 4 (FLUORESCENCIA, ABSORCIÓN Y DIFRACCIÓN DE RAYOS X)
-
COMENTARIOS
Fluorescencia y absorción de rayos X
Dos técnicas análogas a la fluorescencia y absorción atómicas son la fluorescencia y la absorción de rayos X. La diferencia principal es que en rayos X la muestra se excita con radiaciones mucho más energéticas que en espectroscopía atómica.
Cuando se irradia una muestra con fotones de rayos X, parte de estos fotones se absorben y parte se transmiten. La absorción va seguida de emisiones fluorescentes de rayos X. Cada tipo de átomo absorbe rayos X de una forma específica, e igualmente, los reemite por fluorescencia de una forma característica. Dicho de otro modo, cada elemento químico tiene sus propios espectros de absorción y de fluorescencia de rayos X que sirven para diferenciarlo de otros elementos.
La fluorescencia es una técnica esencialmente elemental (es decir, permite identificar elementos químicos como Fe, Co…); la absorción da también información molecular. Una gran ventaja de estas técnicas sobre la espectroscopía atómica es que, a pesar de que las radiaciones son más energéticas, no destruyen la muestra porque no hay que atomizarla previamente. Además, los espectros de fluorescencia de rayos X son mucho más simples que los de fluorescencia atómica. Por su lado, los espectros de absorción de rayos X son muy peculiares: constan de un fondo continuo con picos. Cuando se estudia el espectro en alta resolución se puede observar que estos picos tienen estructura fina cuya inspección es objeto de varias técnicas derivadas.
En general, los átomos pesados absorben más radiación X que los ligeros. Por otro lado, las muestras absorben más cuanto más densas son y absorben menos cuanto más energética (y, por tanto, penetrante) es la radiación. Gran parte de la fluorescencia generada es reabsorbida por la muestra, por lo que la técnica de fluorescencia sobre todo es válida para estudiar la composición de la superficie de las muestras.
Estas técnicas no son especialmente adecuadas para analizar compuestos orgánicos porque estos están compuestos por elementos ligeros, como el C o el N, que absorben poco y consecuentemente dan escasa fluorescencia. Además, esta puede ser absorbida por los elementos pesados de la matriz. Finalmente, en los elementos ligeros la fluorescencia sufre la competencia de un fenómeno llamado de emisión de electrones de Auger, que son electrones arrancados del átomo. No obstante, se pueden determinar elementos de números atómicos muy bajos (entre 5 y 10) en condiciones especiales. Sí es ideal la fluorescencia para analizar metales.
Estas técnicas también se emplean con fines cualitativos, ya que en absorción es aplicable una ley análoga a la de Beer y en fluorescencia la intensidad de la emisión es proporcional a la cantidad de muestra. El problema es que para obtener resultados exactos y repetibles es necesario preparar la muestra muy cuidadosamente. Estas técnicas mejoran mucho cuando se emplea como fuente la radiación X producida por un sincrotrón.
Los espectros de fluorescencia de rayos X se pueden registrar mediante equipos llamados “dispersores de longitudes de onda” o mediante equipos “dispersores de energía” (EDX).. Cada tipo tiene sus ventajas e inconvenientes, pero en general los últimos están ganando terreno en los laboratorios.
Difracción
Otro fenómeno que experimentan los rayos X y que puede ser aprovechado con fines analíticos es su difracción, es decir, el cambio de dirección que experimentan al atravesar la muestra, lo que produce interferencias explicables por la llamada ley de Bragg y que se plasman en el patrón o en el espectro de difracción de rayos X de esa muestra. Este es característico de cada especie química, por lo que puede ser usado en análisis cualitativo.
En análisis suelen ser necesarias cantidades muy pequeñas de muestras y prácticamente sin preparación, si bien se obtienen los mejores resultados triturándola y homogeneizándola hasta obtener un polvo microcristalino. Se pueden identificar tanto átomos como moléculas, e incluso se pueden distinguir distintas fases cristalinas de una misma especia química. No es una técnica adecuada para investigar trazas en sólidos ni tampoco para estudiar líquidos, gases ni, en general, sistemas amorfos.
Espectroscopía de electrones y microscopía de barrido
En absorción y fluorescencia de rayos X se suele excitar la muestra con fotones, para que los absorba, y se observan los fotones emitidos. Pero, en general cuando se irradia una muestra con radiaciones de tan alta energía también se arrancan electrones de la muestra. El análisis espectroscópico de la energía de los electrones arrancados es la base de diversas técnicas conocidas como espectroscopía de electrones (las más impritantes son la fotoelectrónica y la de Auger). Las características de los electrones arrancados también arrojan información topográfica sobre la muestra; este es el fundamento del microscopio de barrido electrónico. Un microscopio de barrido se suele acoplar con un detector de fluorescencia de rayos X para, de esa manera, ver al mismo tiempo la muestra ampliada y analizarla. Es la espectroscopía SEM/EDX.
MATERIAL DIDÁCTICO
«Notas de aplicación» de las espectroscopías de fluorescencia y difracción de rayos X a problemas ambientales:
^^^Volver al índice
Tema 5 (ESPECTROSCOPÍA DE ABSORCIÓN UV-VISIBLE Y DE
LUMINISCENCIA)
-
COMENTARIOS
Espectroscopía de absorción UV-visible
No sólo los átomos pueden absorber energía para después emitirla; también las moléculas (conjuntos de átomos) lo hacen. Muchas moléculas se excitan cuando se irradian con fotones de diversa energía. En este tema se estudia cómo lo hacen cuando se irradian con luz o con fotones ultravioleta, y el partido que de ello puede sacarse. (Este fenómeno, por cierto, justifica en muchos casos el color que presentan muchas sustancias químicas, color que es el complementario del de la radiación UV-visible absorbida.) Después de la excitación las moléculas se relajan, entre otros modos emitiendo luz fluorescente o fosforescente. Esa es la base de la espectroscopía de luminiscencia.
Los espectros de absorción UV-visible no están formados de picos como en espectroscopía atómica o en fluorescencia de rayos X, sino de bandas más o menos anchas. La anchura de estas señales, su posición en el espectro, su intensidad e incluso su número dependen de diversos factores, como el estado físico (más estrechas en gases), la naturaleza del disolvente, la temperatura, el pH… El hecho de que aparezcan pocas bandas y que sean tan dependientes de variables externas hace que esta técnica sea poco adecuada para la identificación, si bien sí suele ser posible adscribir la especie a la familia química correspondiente o averiguar la presencia en la molécula de grupos químicos absorbentes o cromóforos.
Sin embargo, la espectroscopía de absorción UV-visible sí es muy apropiada para realizar análisis cuantitativos una vez conocida la naturaleza de la muestra, ya que la ley de Beer se cumple muy bien. Incluso puede aplicarse normalmente la ley de aditividad de absorbancias, según la cual la absorbancia de una mezcla es la suma de la absorbancias de los componentes individuales.
La técnica es idónea para compuestos orgánicos líquidos, gaseosos o en disolución que poseen instauraciones (enlaces múltiples); para iones de elementos de transición, libres o compuestos (Cr3+, MnO4-…) y para compuestos organometálicos de lantánidos y actínicos. También es aplicable a compuestos no absorbentes de radiación UV-visible si son capaces de unirse a un reactivo de modo que se forme un compuesto absorbente. La técnica permite, por otro lado, detectar fotométricamente el punto de equivalencia en muchas valoraciones.
Los aparatos para espectroscopía UV-visible constan de una fuente, un monocromador y un detector y suelen ser relativamente sencillos y baratos en comparación con los instrumentos requeridos en otras técnicas.
Espectroscopía de luminiscencia
Tras la absorción de radiación UV-visible las moléculas pueden relajarse de modo no radiante (calor) o radiante; en este caso pueden hacerlo de dos modos: por fluorescencia y por fosforescencia, formas ambas de fotoluminiscencia. La primera técnica se emplea mucho más. Aunque no es muy apropiada para identificar sustancias, sí lo es para cuantificarlas (incluso es mejor que la absorción UV-visible) porque la señal es lineal con la concentración en un intervalo bastante ancho y los límites de detección son muy bajos. El inconveniente es que sólo una pequeña parte de los compuestos orgánicos son luminiscentes. Además, los efectos de matriz son considerables. En general, hay que prestar atención a todas las causas que puedan producir quenching (desactivación de la fotoluminiscencia).
En estas técnicas se trabaja con dos tipos de espectros, el llamado de excitación (esencialmente igual al de absorción) y el de emisión. Los mejores fluorímetros son los que pueden registrar ambos. Hoy día estas técnicas se han implementado en pequeños dispositivos llamados sensores que miden automáticamente la concentración de uno o varios analitos (por ejemplo, el sensor de fluorescencia de oxígeno).
En medio ambiente las técnicas fotoluminiscentes permiten estudiar especies de interés ambiental tanto líquidas como sólidas y gaseosas. Se han adaptado sondas de fluorescencia a penetrómetros para estudiar subsuelos.
Relacionada con la fotoluminiscencia está la técnica de la quimioluminiscencia, consistente en medir la luminiscencia producida en una reacción química. También se pueden hacer análisis inmunoquímicos (reacción de un antígeno con un anticuerpo) con ayuda de la fotoluminiscencia.
Técnicas relacionadas
La turbidimetría, la nefelometría, la refractometría, la espectrometría fotoacústica, la polarimetría, la dispersión óptica rotatoria o el dicroísmo circular son otras técnicas en las que se hace uso de la luz visible (o ultravioleta o infrarroja) para estudiar la materia.
MATERIAL DIDÁCTICO
«Notas de aplicación» de las espectroscopías UV-visible y de fluorescencia a problemas ambientales:
^^^Volver al índice
Tema 6 (ESPECTROSCOPÍAS INFRARROJA Y RAMAN)
-
COMENTARIOS
Espectroscopía de absorción IR
En las moléculas, los átomos vibran según modos normales de vibración característicos con frecuencias de vibración también características de estos modos. Estas frecuencias son del orden de las que poseen los fotones de la radiación infrarroja. Cuando una molécula se irradia con estos fotones, es capaz de absorberlos. Pero solo se absorben aquellos cuyas frecuencias coinciden exactamente con las de los modos de vibración de esa molécula concreta (siempre que se cumplan ciertos requerimientos físicos adicionales). En este fenómeno se fundamenta la espectroscopía de absorción infrarroja, que tiene como correlata a la espectroscopía de emisión IR, si bien esta apenas se utiliza.
Aunque la espectroscopía de absorción IR permite cuantificar especies (ya que le es aplicable la ley de Beer), su mayor potencia radica en la posibilidad de identificar especies químicas. Los espectros están constituidos por bandas (generalmente anchas, excepto en gases simples, cuyos espectros están formados más bien de picos), cada una de las cuales corresponde, en general, a la vibración de un grupo químico determinado de la molécula (-NH2, =C=O, -CH2-, etc.). Consultando tablas que recogen las frecuencias características aproximadas de estos grupos, estos se pueden identificar. (Por ejemplo, las moléculas que contienen un grupo C=O presentan una banda en torno a 1650 cm-1.) De este modo, puede reconstruirse la estructura de la molécula (a modo de un rompecabezas). A veces esta técnica no es suficiente para identificar la molécula, pero con la ayuda de la resonancia magnética nuclear (tema 7) y la espectrometría de masas (tema 8), la identificación suele resultar infalible.
La técnica de absorción IR puede aplicarse a sustancias sólidas, líquidas y gaseosas. Los sólidos se suelen dispersar en una pastilla de KBr o en una parafina líquida llamada nujol. Los líquidos y gases se estudian en células entre «ventanas» transparentes a la radiación IR. Los líquidos han de disponerse en películas muy finas para evitar la absorción total. Es complicado estudiar disoluciones acuosas porque el agua absorbe mucha radiación IR y supone una seria interferencia para reconocer las especies disueltas. También interfieren los picos del agua atmosférica, e incluso el CO2 atmosférico. Por ello, al igual que en espectroscopía UV, es conveniente restar el espectro del disolvente del de la disolución. En muchos casos, operando así se evitan las interferencias espectrales de dicho disolvente. De cualquier modo, siempre se obtienen mejores resultados si se opera en vacío o bien en una atmósfera de un gas no activo –que no da bandas– en infrarrojo como el N2.
La espectroscopía IR tiene bastantes aplicaciones en medio ambiente. Una de ellas es estudiar la contaminación urbana mediante un espectrómetro situado en un edificio que emite radiación y la recibe tras reflejarse esta en un espejo situado en otro edificio, midiéndose el tipo y la cantidad de radiación absorbida. En general, se pueden estudiar todo tipo de sustancias con enlaces covalentes. Aquellas formadas exclusivamente por enlaces iónicos son más difíciles de observar en IR.
Los espectrómetros clásicos (llamados dispersivos) están formados por una fuente, un monocromador y un detector. Los hay de un solo haz y de haz doble. Los modernos, llamados interferómetros, están basados en el fenómeno físico de las interferencias y en la operación matemática de la transformación de Fourier. Además de no necesitar monocromador son muchísimo más rápidos (en menos de un segundo se registra un espectro), tienen más resolución y la relación señal/ruido es mucho mejor. Por ello, los interferómetros (que no necesitan monocromador) han desplazado completamente a los espectrómetros dispersivos.
Se puede acoplar un microscopio óptico a un espectrómetro IR para focalizar la radiación IR sobre puntos muy pequeños de una muestra y analizar la radiación refleja por esta.
Una técnica derivada es la espectroscopía de absorción en el infrarrojo próximo, muy empleada en análisis rutinarios de plásticos y agroalimentarios.
Espectroscopía Raman
Una técnica estrechamente relacionada con la IR pero que descansa en bases físicas diferentes es la espectroscopía Raman, así llamada en honor a su descubridor. Cuando una muestra se irradia monocromáticamente con fotones visibles, UV o IR (normalmente generados por un láser) se observa que una parte de la radiación se dispersa (cambia de dirección) en el espacio. Lo más interesante es que algunos de los rayos salen dispersados con frecuencias distintas de la de los rayos incidentes y que las diferencias de frecuencias coinciden con las frecuencias de las bandas del espectro IR. Por ello, el espectro Raman es bastante parecido al infrarrojo, si bien hay bandas que aparecen en uno y no en otro. Las técnicas son, pues complementarias.
Una desventaja del Raman es que el efecto es muy débil (si bien en algunos casos se puede amplificar (por ejemplo, adsorbiendo los analitos sobre superficies, lo que da lugar a la técnica derivada llamada SERS). Por ello, los límites de detección son más bien altos (ello se ha contrarrestado en la actualidad usando detectores extraordinariamente sensibles; tanto, que incluso recogen fotones del ambiente, por lo que los espectros Raman a menudo se registran en completa oscuridad). Una gran ventaja es que el agua da pocas bandas en Raman, y estrechas, por lo cual las interferencias del disolvente en disoluciones acuosas es escasa. Tampoco interfieren seriamente los gases atmosféricos. Por todo ello, los requerimientos de preparación de la muestra suelen ser mucho menores que en IR. Los espectros se interpretan como los IR: por medio de tablas de frecuencias características de grupos químicos.
Existen dos tipos de equipos: los clásicos y los basados en la transformación de Fourier. Sin embargo, en Raman, a diferencia del IR, no está tan claro que los segundos sean más ventajosos que los primeros. Si mejoran mucho el espectro en caso de que se produzca en exceso el fenómeno de la fluorescencia (tema 5), que suele competir con el Raman, suponiendo una seria interferencia.
La técnica, como la IR, también se puede implementar en un microscopio, con excelentes resultados.
En medio ambiente las aplicaciones son numerosas. Una nueva ventaja sobre la espectroscopía IR es que como la fuente de excitación usual es luz visible, esta puede conducirse mediante una fibra óptica donde se quiera. Esto permite llevar la radiación al subsuelo mediante penetrómetros y traer de vuelta la radiación Raman dispersada, hacia el detector.
Técnicas relacionadas
Una técnica directamente relacionada con las vibracionales es la espectroscopía rotacional. Con radiación de microondas se puede excitar la muestra de modo que cambie exclusivamente el estado de rotación (no de vibración) de sus moléculas. Se pueden estudiar así gases atmosféricos contaminantes como el CO. El lidar, una técnica derivada, permite estudiar aerosoles y estratificaciones de polvo atmosférico.
MATERIAL DIDÁCTICO
«Notas de aplicación» de las espectroscopías IR y Raman a problemas ambientales:
^^^Volver al índice
Tema 7 (RESONANCIA MAGNÉTICA NUCLEAR)
-
COMENTARIOS
Teoría
Los núcleos de los átomos pueden considerarse diminutos imanes. Y como todos los imanes, se orientan cuando se ponen en el seno de un campo magnético externo. Por ejemplo, dado un conjunto de protones (1H), unos se orientan en la misma dirección que el campo y otros en contra. Ambos tipos de protones tienen distinta energía, pero la diferencia es muy pequeña (del orden de la de las radiofrecuencias). Cuando se irradia con radiofrecuencias esa colección de protones, los de menor energía pueden absorber fotones y pasar al estado de energía superior. En esto se basa la técnica de la Resonancia Magnética Nuclear (RMN).
Es una técnica, en general, unielemental en el sentido de que en cada experimento solo se estudia un tipo de núcleo (por ejemplo, en un experimento se estudian solo los protones, pero no los carbonos o los oxígenos de la molécula). Hay que tener en cuenta también que no todos los núcleos pueden ser estudiados por RMN. Un ejemplo muy importante de núcleo que no es activo en RMN es el de 12C, si bien sí lo es el de su isótopo 13C.
Hay que insistir en que la división de los núcleos de un conjunto en distintos niveles de energía no preexiste, sino que es provocada por un campo magnético exterior. Cuanto mas intenso es ese campo, mayor es la diferencia de energía entre los niveles provocados y más bajo es el límite de detección de la técnica. Por otro lado, para un mismo valor del campo magnético aplicado, los niveles de energía de los núcleos de los distintos elementos químicos experimentan diferentes magnitudes de desdoblamiento. Por tanto, cada elemento químico absorbe fotones de diferente frecuencia. En esto radica la especificidad de la técnica.
Pero es más: no todos los núcleos de un mismo elemento absorben fotones de exactamente la misma frecuencia. Eso depende de la posición de cada núcleo en la molécula; de su entorno químico (es decir, de la naturaleza, posición y número de los núcleos del mismo o distinto elemento que lo rodean). Esto permite distinguir núcleos de un elemento determinado según el grupo funcional en que se integren. Por ejemplo, se pueden distinguir los protones de un grupo –CH3 (metilo) de los de un grupo –OH (alcohol), -CHO (aldehído) o -COOH.(ácido carboxílico). Cada uno de estos tipos de protones da, en principio, una señal propia en el espectro de RMN.
La frecuencia de cada señal se suele medir en relación a la de otra que se toma como referencia y a la que se le asigna el valor 0. La diferencia relativa de frecuencias entre una señal dada y la que se ha adoptado como referencia se llama desplazamiento químico. Por otra parte, el área de cada señal es proporcional al número de protones que representa. Así, el área de la señal de los protones del grupo -CH3 es triple de la del protón de –COOH.
Con un instrumento de alta resolución se puede apreciar que, en realidad, cada una de estas señales suele estar desdoblada formando dobletes, tripletes, cuadrupletes, etc. Ello se debe a los diminutos campos magnéticos de los núcleos próximos a uno dado, que modifican levemente la intensidad del campo magnético externo. Este efecto, llamado acoplamiento de espines, complica el espectro pero, en cambio, ayuda a identificar el tipo de núcleos representado por la señal y también la naturaleza de los grupos vecinos. La separación entre los picos de estas señales múltiples se llama constante de acoplamiento y proporciona información fundamental.
Midiendo las constantes de acoplamiento típicas de las distintas señales de muchas moléculas se ha comprobado que sus valores responden a patrones muy reproducibles. Esto ha permitido construir tablas que “predicen” la constantes de acoplamiento de las señales de cualquier molécula en estudio. Igualmente, se han tabulado desplazamientos químicos típicos de los distintos grupos funcionales incluso teniendo en cuenta la influencia de los grupos vecinos. Estas tablas son mucho más precisas que las análogas de frecuencias características en IR o UV. Las tablas de correlación de RMN son fundamentales para la interpretación de cualquier espectro.
Práctica
Por todo lo expuesto, la técnica de RMN tiene extraordinarias aplicaciones en la identificación de sustancias desconocidas más que en su cuantificación, ya que los límites de detección son altos. Se suele decir que el RMN, la espectroscopía IR y la espectrometría de masas forman un trío de técnicas complementarias suficiente para averiguar la naturaleza y estructura de cualquier compuesto químico desconocido. La RMN es muy usada en los laboratorios de química orgánica. La variedad más empleada es la RMN de protones, seguida de la de carbono-13. También son muy útiles las de nitrógeno y fósforo-31.
Las muestras más fácilmente abordables por RMN son las líquidas y las que se encuentran disueltas (si se van a estudiar protones, los de la molécula de agua interfieren, por lo que la disolución ha de hacerse en agua deuterada). La técnica también se puede aplicar a sólidos, aunque los espectros son más difíciles de obtener.
Antiguamente la técnica de RMN se practicaba en la modalidad llamada de onda continua. Hoy ha sido reemplazada muy ventajosamente por la Resonancia Magnética Nuclear de Transformada de Fourier, entre otras razones porque esta es muchísimo más rápida (si bien los aparatos son complicados de uso y mantenimiento y muy costosos). En esta variedad los núcleos se irradian con pulsos policromáticos de radiofrecuencia. Se han ideado multitud de secuencias de pulsos para disminuir los límites de detección de determinados núcleos poco sensibles, intensificando sus señales, y también para simplificar los espectros e interpretarlos mejor. Además, el estudio de ciertos efectos como el Overhauser ayudan a resolver la estructura de moléculas tan complejas como los ácidos nucleicos y las proteínas.
Ciertas variedades de la técnica mucho más sencillas pero que han encontrado grandes aplicaciones en medio ambiente se basan en estudiar el tiempo que los núcleos tardan en relajarse después de la excitación, o bien las velocidades de relajación (relaxometría). Permiten estudiar, por ejemplo, la porosidad de las rocas y los fluidos que rellenan estos poros. También es muy útil en medio ambiente la Resonancia Magnética Nuclear de Campo Terrestre, que saca partido del débil aunque extraordinariamente homogéneo campo magnético del planeta para estudiar rocas, hielo, yacimientos geológicos, etc.
Una técnica relacionada con la RMN es la resonancia de espín electrónico, basada en excitar magnéticamente a los electrones, no a los núcleos. Una aplicación importante en medio ambiente es el estudio de los muy dañinos radicales libres.
MATERIAL DIDÁCTICO
Aplicaciones de la espectroscopía RMN a problemas ambientales:
^^^Volver al índice
Tema 8 (ESPECTROMETRÍA DE MASAS)
-
COMENTARIOS
Espectroscopia atómica de masas
Hasta aquí se han considerado distintas técnicas instrumentales con un aspecto en común: los instrumentos producen espectros, que son una representación gráfica de la frecuencia de los fotones (absorbidos, transmitidos, dispersados…) frente a su intensidad. Por analogía, una representación gráfica de las masas atómicas o moleculares de los elementos o moléculas que forman una muestra frente a su abundancia se llama un espectro de masas. La técnica que los produce es la espectrometría de masas y cabe distinguir la espectrometría atómica de masas de la molecular.
La técnica se basa en el hecho conocido de que una partícula cargada que se mueve rectilíneamente y penetra en un campo eléctrico o magnético experimenta una deflexión; es decir, su trayectoria se curva. Y lo hace más pronunciadamente cuanto menos pesa. De este modo, si se dispone de un conjunto de iones en fase gaseosa y se dirigen hacia el interior de un campo eléctrico o magnético, experimentarán distintos grados de deflexión según su masa iónica. Si a la salida del campo se coloca un detector adecuado, este podrá «contar» los iones de cada masa que llegan a él y reflejarlo en el espectro de masas de esa muestra.
Lo primero que hay que conseguir es poner la muestra en fase gaseosa e ionizarla. Para ello se pueden emplear distintas fuentes de excitación energética. Cuando se usa un plasma, la excitación es tan alta que la molécula se parte completamente, es decir, da lugar a los átomos que la constituyen, los cuales al mismo tiempo quedan ionizados. Lo que se obtiene con ello es un espectro atómico de masas. Su interpretación es trivial: cada haz de iones de determinada masa que el espectrómetro separa espacialmente da un pico propio. La identificación es inmediata porque la masa del ion es su masa atómica; basta consultar una tabla periódica. Por supuesto, son posibles las interferencias ya que hay isótopos de algunos elementos que pesan lo mismo que los isótopos de otros. No obstante, como se conocen las abundancias relativas naturales de los isótopos de todos los elementos químicos, es relativamente sencillo –el mismo software con que se controla el equipo lo hace– detectar estas interferencias. (Por ejemplo, si la molécula tiene átomos de Cl, deben aparecer en el espectro dos picos debidos a 35 y 37 unidades de masa con una relación de intensidades aproximadamente 3:1. Si la relación medida es diferente es que uno de los picos sufre una interferencia de otro elemento químico también presente.
Espectroscopia molecular de masas
Si se emplea una fuente menos energética que el plasma la molécula probablemente no se partirá completamente en sus átomos, sino en fragmentos más o menos grandes. Eso depende del método de fragmentación empleado. Así, hay métodos tan «suaves» que ni siquiera la parten, pero sí la ionizan; otros, en cambio, pueden fragmentarla considerablemente. Los primeros se denominan de desorción (electrospray, desorción-ionización por láser, bombardeo con átomos rápidos, desorción por campo…). Los segundos se llaman de volatilización o de fase gaseosa (por impacto electrócio, ionización química, ionización por campo…). Ambas estrategias son útiles. Si la molécula no se fragmenta en absoluto (solo se ioniza), se puede determinar su peso molecular; y si lo hace, se puede determinar el peso de esos fragmentos, lo que ayuda a la identificación. Por ejemplo, si al romper una molécula orgánica se obtienen trozos de 14 unidades de masa, probablemente corresponden a grupos -CH2-; si se obtienen trozos de 18 y 31 unidades de masas podría pensarse en un alcohol porque se sabe que este tipo de moléculas suele conducir a la producción de moléculas de agua ionizadas y al fragmento CH2OH+.
Existen mucha información experimental recabada tras décadas de aplicación de la técnica que permiten interpretar un espectro molecular de masas y conocer, por los fragmentos obtenidos, de qué molécula se trata. La experiencia indica cuáles son los patrones de fragmentación más comunes en los distintos tipos químicos de moléculas (alcoholes, aldehídos, cetonas, ácidos, ésteres, éteres…). En cierto modo, se trabaja como cuando se arma un rompecabezas, si bien actualmente es el software que llevan incorporado estos equipos quien recompone la molécula a partir de los pesos de los fragmentos detectados y de las relaciones isotópicas conocidas. Por ello, esta técnica es, junto a la infrarroja y a la resonancia magnética molecular, de las más potentes para identificar una especie desconocida.
Los equipos par realizar espectrometría de masas son técnicamente complejos y, por tanto, caros. En el interior de ellos se hace el vacío para que los iones no impacten contra moléculas de aire que desvíen su trayectoria. El elemento que hace las veces de «monocromador» en esta técnica, es decir, el que separa los iones según su masa para hacerlos llegar al detector, es el llamado analizador de masas. Los analizadores de masas más usados son el cuadrupolo, la trampa de iones, el analizador de tiempo de vuelo y el de sector magnético. Modernamente también se emplean analizadores basados en las transformación de Fourier, que permiten obtener espectros de extraordinaria resolución con alta relación señal/ruido.
Son muy útiles para analizar mezclas los espectrómetros de masas en tándem. Se trata de dos equipos colocados en serie. El primero aplica un método de fragmentación suave de modo que sólo ioniza las moléculas. El segundo va recogiendo uno a uno los haces de las distintas moléculas separadas, las cuales fragmenta más enérgicamente para así identificarlas a partir de los trozos obtenidos.
La cuantificación mediante espectrometría de masas se realiza mediante el uso de patrones de los que, como de costumbre, se obtiene también el espectro en las mismas condiciones que el de la muestra. Conviene aplicar el método del patrón interno. En espectroscopía atómica de masas da muy buenos resultados el método de la dilución isotópica, consistente en añadir a la muestra un patrón enriquecido en uno de los isótopos del analito. Obtenidos los espectros de la muestra y de la muestra con el patrón enriquecido, la aplicación de ciertas fórmulas matemáticas basadas en los datos conocidos de abundancia natural de los isótopos permite cuantificar con poco error el analito.
La espectrometría de masas es una técnica también útil para analizar la composición química de la superficie de un material. Por otro lado, existen sensores capaces de adsorber gases y determinar la masa de gas adsorbido.
En medio ambiente la espectrometría de masas tiene múltiples aplicaciones ya que, como se ha dicho, pueden determinarse isótopos de un mismo elemento. Esto permite, por ejemplo, distinguir los radiactivos de los no radiactivos. Además, las proporciones isotópicas de muchos elementos dan información ambiental muy interesante. Por ejemplo, la relación 13C/12C permite estimar la incorporación de nueva materia orgánica al suelo. Existen espectrómetros de masas que pueden realizar los análisis in situ.
MATERIAL DIDÁCTICO
Ejemplos de aplicación de la espectrometría de masas a problemas ambientales:
^^^Volver al índice
Tema 9 (POTENCIOMETRÍA)
-
COMENTARIOS
Pilas electroquímicas
Las distintas especies químicas tienen diferente afinidad por los electrones; es decir, unas los captan con más facilidad que otras. Cuando una sustancia capta electrones se dice que se reduce. Las que tienen mucha afinidad por los electrones se dice que tienen alto potencial de reducción, que es una magnitud que se mide en voltios. Cuando se ponen en contacto dos especies químicas, en general la que tiene más alto potencial de reducción toma electrones de la que lo tiene más bajo. Esta última se dice que se oxida. En esto se basan las pilas electroquímicas.
Una pila o celda electroquímica consta en general de dos semipilas o semiceldas. En una, llamada catódica, se produce una reacción de reducción y en la otra (anódica) una de oxidación. Los electrones que se liberan en una de ellas se pueden hacer circular por un conductor para que lleguen a la otra. Si en el conductor se interpone una bombilla, esta luce. Para que el circuito quede convenientemente cerrado hay que comunicar ambas semiceldas mediante algún sistema conductor adecuado, normalmente un puente salino (una disolución de una sal).
Cada pila genera un potencial cuyo valor depende de la naturaleza química de las especies que forman las semipilas y de su concentración. La llamada ecuación de Nernst liga estas dos variables, y también la temperatura, con el potencial. Cuando la concentración (o más propiamente la actividad) de todas las especies electroactivas es la unidad, el potencial de la pila se llama potencial normal.
El potencial normal de cada pila se puede calcular fácilmente restando el potencial normal de la semipila catódica menos el de la anódica: E = Ecát – Eán. Los potenciales normales de cada semipila se pueden encontrar en tablas. Se han podido conocer enfrentando cada semipila a una semipila normal de hidrógeno o electrodo normal de hidrógeno –los términos semipila y electrodo se suelen emplear indistintamente-. Un electrodo normal de hidrógeno está formado por un alambre de platino introducido en una disolución que contiene protones (H+) en concentración unidad en la que se hace burbujear gas hidrógeno a 1 atm de presión. El potencial de una semipila no se puede medir en términos absolutos. Al electrodo normal de hidrógeno se le asigna arbitrariamente un potencial de 0 voltios. De este modo, midiendo el potencial global de la pila construida y sabiendo que el de la semipila de hidrógeno es 0 (independientemente de que actúe como cátodo o como ánodo), se puede calcular el potencial de cualquier semipila enfrentada a él a partir de la mencionada expresión E = Ecát – Eán. Este potencial de semipila es el que se tabula.
Potenciometría
Como se ha dicho, el potencial de una pila depende de la naturaleza química de las especies que la forman y de su concentración. En esto reside precisamente la capacidad analítica de la técnica potenciométrica, cuyas aplicaciones, en general, son cuantitativas más que cualitativas. Es decir, no se suele emplear para identificar especies, pero sí para medir la concentración de especies cuya naturaleza ya se conoce.
Los equipos para llevar a cabo la potenciometría son muy sencillos y baratos. Básicamente consisten en dos recipientes, un puente salino y dos electrodos, que normalmente son piezas de un metal u otra especie conductora adecuada (a menudo, grafito). Estos se introducen en las disoluciones de ambas semipilas. Ambas semipilas se suelen integrar en un solo módulo para hacer el sistema más manejable. Dentro del módulo, el punte salino se suele reemplazar por una membrana permeable a algunos iones.
Se han clasificado los electrodos o semipilas en tres categorías: de primera, segunda y tercera clase. Los de primera clase están formados por un metal introducido en una disolución de una sal del mismo metal. Son útiles para determinar la cantidad de ese metal en disolución a partir de la ecuación de Nernst. Ahora bien, esto se puede hacer siempre que se conozca el potencial global de la pila (es fácil: se mide con un potenciómetro) y el potencial de la otra semipila. En la práctica, la otra semipila suele ser una cuyo potencial es bien conocido y estable. Por eso, este electrodo se llama de referencia, mientras que el de la disolución cuya concentración se quiere medir se llama indicador. Se han ideado varias electrodos de referencia. El más universal es el electrodo normal de hidrógeno, ya comentado. Pero por su difícil uso, en la práctica se emplean otros electrodos de referencia como el de calomelanos o el de Ag/AgCl.
Además de estos electrodos, existen otros más evolucionados que se suelen denominar sensores potenciométricos. Los sensores potenciométricos más comunes son los llamados electrodos de membrana, cuyo nombre responde a que se introducen en la disolución cuya concentración en una especie electroactiva se quiere medir y quedan separados de esta por una membrana especialmente fabricada. Esta membrana es hasta cierto punto permeable a los iones que se quieren determinar. Por eso, a estos electrodos también se les da el nombre de electrodos selectivos de iones.
El electrodo de membrana más conocido es el llamado de vidrio o electrodo de pH. Su membrana es sensible a los protones del medio, por lo que con dicho electrodo se puede medir el pH de una disolución. Para hacerlo hay que calibrar previamente el electrodo con tampones de pH conocido. El general, las curvas de calibración son rectas a valores de pH no demasiado bajos o demasiado altos. A pHs altos se suele cometer el llamado error alcalino (debido, sobre todo, a iones alcalinos que interfieren con los protones); a pHs muy bajos, el error ácido.
En general, mediante un electrodo selectivo de un ion determinado I se puede determinar el pI de la disolución (pI = -log[I]). Con estos electrodos se puede medir rápidamente la concentración de iones como Cl-, Br-, I-, F-, CN-, SCN-, NO3-, S=, Ag+, Cd++, Ca++, Pb++, etc. Otros sensores potenciométricos son los llamados transistores de efecto de campo. También existen sensores potenciométricos de moléculas (no de iones). Basados en reacciones bioquímicas, se puede medir con ellos concentraciones de gases como CO2, NH3 H2S o SO2 y de biomoléculas.
Por todo lo explicado, se comprenderá que los equipos de medida potenciométrica tienen grandes utilidades en medio ambiente. Permiten medir fácilmente el pH, la alcalinidad de las aguas, su dureza y su potencial redox y además determinar metales y gases disueltos.
Otra utilidad de la potenciometría son las valoraciones potenciométricas, que son aquellas en que el punto de equivalencia se detecta haciendo medidas del potencial del sistema. Tienen grandes ventajas sobre las valoraciones basadas en la observación de un cambio de color o de la aparición de un precipitado, las cuales son muy subjetivas.
MATERIAL DIDÁCTICO
Ejemplos de aplicación de la potenciometría a problemas ambientales:
^^^Volver al índice
Tema 10 (TÉCNICAS DE CORRIENTE ELÉCTRICA)
-
COMENTARIOS
Electrolisis
En el tema anterior se estudió cómo las reacciones químicas llamadas de oxidación- reducción (redox) producen corriente eléctrica. Pues bien, a la inversa, la corriente eléctrica también puede propiciar que se produzcan reacciones químicas. Cuando se hace pasar electricidad por una celda electroquímica como las estudiadas en el tema anterior, se suele producir una ruptura en las especies en disolución (electrolisis), formándose nuevas especies. Para que se produzca la electrolisis, la fuente de corriente tiene que tener un potencial mayor y de sentido contrario al de la pila correspondiente.
En este fenómeno se basan distintas técnicas eléctricas como la electrogravimetría, la culombimetría, la conductimetría y la voltamperometría (hay autores que prefieren distinguir entre voltametría y amperometría). La diferencia sustancial de estas técnicas con la potenciometría es que en esta última se actúa procurando que apenas pase corriente por el sistema, mientras que en aquellas el paso de corriente es precisamente lo que permite estudiar el sistema.
Elecrogravimetría y culombimetría
Cuando como consecuencia de la electrolisis un ion metálico de la disolución (por ejemplo Cu++) de reduce y convierte en metal, este metal normalmente se puede pesar (bien porque queda adherido al electrodo o porque precipita y se puede recuperar, lavar y secar). De este modo, esperando a que la reacción se complete se puede, por simple pesada, saber cuánto metal iónico (Cu++ en este ejemplo) había en disolución. Esta técnica se denomina por ello electrogravimetría. Una alternativa es medir la carga que ha de pasar por el circuito para que todo el analito se transforme (en el ejemplo, para que todo el Cu++ pase a Cu). Como la carga necesaria es proporcional a la cantidad de analito, esta otra técnica también permite medir concentraciones. Se llama, por motivos obvios, culombimetría.
Es preciso comentar algunos aspectos prácticos de estas técnicas. Cuando se mantiene constante el potencial externo aplicado, la corriente que pasa por el circuito va disminuyendo con el tiempo porque cada vez habrá menos electrolitos capaces de transportar la cargar (ya que están desapareciendo debido a la reacción electroquímica). Eso provoca que la electrolisis se ralentice. Si para evitarlo se va aumenta el potencial conforme pasa el tiempo se corre el riesgo de que también se electrolicen especies distintas del analito de interés; es decir, que se produzcan interferencias (salvo que la especie de interés sea la única que existe en disolución). Como se ve, ambos procedimientos tienen sus ventajas e inconvenientes. Según el caso, se opta por el que tenga menos inconvenientes.
Cuando se procura mantener el potencial constante, la técnica (ya sea electrogravimétrica o culombimétrica) se llama potenciostática; si se lleva a cabo a intensidad de corriente constante, se denomina galvanostática. La modalidad potenciostática se realiza con ayuda de un potenciostato, que es un aparato constituido por tres electrodos: ánodo, cátodo –uno de ellos se llama electrodo de trabajo y el otro electrodo auxiliar, según los casos– y un electrodo de referencia que se conecta al de trabajo y cuya función es mantener constante el potencial de este para evitar interferencias. Existen equipos llamados potenciostatos-galvanostatos que permiten realizar indistintamente ambas variedades de estas técnicas.
Una aplicación muy interesante de la culombimetría es la valoración culombimétrica. En ella, a partir de una sustancia química precursora se produce electrolíticamente el agente valorante, el cual va reaccionando con el analito hasta que este se consume. Si se continúa realizando la electrolisis, el agente valorante quedará en exceso y podrá detectarse mediante un indicador o bien potenciométrica o conductimétricamente. Esto marca el final de la valoración.
Voltamperometría
Cuando se hace pasar corriente eléctrica por un circuito, la intensidad es proporcional al potencial de la fuente externa. Para determinados valores del potencial, el cambio de la intensidad con el potencial es lineal según predice la ley de Ohm: V = IR, siendo R la resistencia, pero a partir de ciertos valores del potencial la recta se curva hasta perder toda su pendiente. Se dice que se ha alcanzado la corriente límite. A partir de ese momento, aunque se aumente el potencial la corriente no aumentará. Esto es debido a distintos fenómenos que ocurren en el sistema y que se engloban bajo el nombre de polarización. Por ejemplo, los iones y electrones que se mueven hacia o desde los electrodos lo hacen con una velocidad que no puede superar ciertos límites porque se producen impedimentos y choques; además, las especies emplean cierto tiempo en adsorberse o desorberse de los electrodos, en cristalizar, etc.
La polarización tiene otra consecuencia: que para que se inicie la electrolisis hay que proporcionar un potencial externo superior al teórico deducido por la ley de Nernst. Ese exceso se llama sobrepotencial. El sobrepotencial suele ser alto cuando se emplean ciertos electrodos como el de mercurio, cuando los electrodos son muy pequeños o cuando se generan gases en las reacciones electroquímicas que se dan. Ahora bien, tanto la polarización como el sobrepotencial no son inconvenientes en la técnica voltamperométrica. Al contrario, precisamente se basa en dichos fenómenos. Por ejemplo, el valor de la corriente límite depende de la concentración de la especie electroactiva. De ahí una de las utilidades analíticas de la técnica, que se lleva a cabo empleando patrones.
En general, la voltamperometría consiste en estudiar cómo varía la corriente cuando se va cambiando el potencial. La curva correspondiente es el voltamperograma, que tiene forma matemática de una sigmoide llamada onda voltamperométrica. El punto de inflexión de la sigmoide es la corriente límite dividida por 2. El potencial correspondiente a ese punto se denomina potencial de semionda y está relacionado con el potencial de reducción de la especie correspondiente, lo que sirve de base a las posibilidades identificativas de esta técnica. Se pueden determinar varias especies mezcladas si sus potenciales de reducción son suficientemente diferentes. En ese caso, el voltamperograma muestra varias ondas voltamperométricas, una por cada especie. El instrumento para aplicar la técnica es relativamente simple. Consta de tres electrodos: el de trabajo (que debe ser fácilmente polarizable), el auxiliar y el de referencia.
La técnica tiene interesantes variedades, según cómo se modifique el potencial con el tiempo. Así, en la voltamperometría de barrido lineal el potencial se aumenta linealmente con el tiempo; en la cíclica, el potencial se aumenta linealmente para después disminuir, y así en varios ciclos. Los voltamperogramas que se obtienen proporcionan interesantes parámetros útiles para reconocer el analito y medir su concentración.
Una técnica voltamperométrica muy especial es la polarografía, que fue la primera que se desarrolló y una de las que siguen ofreciendo más posibilidades. Su principal característica es que el electrodo de trabajo es una gota de mercurio que se va formando y cayendo continuamente (esto provoca que los polarogramas tengan una peculiar forma de diente de sierra). Se consiguen elevados sobrepotenciales y el electrodo, al renovarse continuamente, siempre está limpio.
Un refinamiento de esta técnicas es la voltamperometría (o polarografía) de pulsos, así llamada porque el potencial se aplica según pulsos (subidas bruscas) programados. Dentro de ellas cabe destacar las subvariedades de voltamperometría diferencial de pulsos y voltamperometría de onda cuadrada. Los voltamperogramas que se obtienen están formados de picos (no de ondas voltamperométricas), por lo que son muy fáciles de interpretar y de ellos se obtiene de forma relativamente sencilla información cualitativa y cualitativa incluso de mezclas, con límites de detección muy bajos.
También produce excelentes resultados otra modalidad llamada voltamperometría de redisolución, consistente en iniciar una electrolisis para que se deposite una especie en el electrodo y después bajar el potencial para dejar que el proceso electroquímico se invierta y el depósito se redisuelva. Lo que se consigue así es una preconcentración del analito, y de esto modo los límites de detección bajan extraordinariamente (hasta 10^(-12 M)). Los voltamperogramas están formados de picos, uno por cada especie existente en la muestra.
Existen sensores basados en las técnicas voltamperométricas. Dos muy empleados son el de glucosa y el de oxígeno. Precisamente la determinación de este gas disuelto es una de las aplicaciones más importantes de la voltamperometría en medio ambiente, junto a la determinación de metales en disolución y, por polarografía, de ciertos compuestos orgánicos.
Conductimetría
Otra técnica electroquímica basada en el paso de corriente eléctrica a través de una disolución es la conductimetría. Se basa en la medida de la conductividad de la disolución, es decir, la facilidad con que fluye la corriente eléctrica a través de ella. La conductividad depende fuertemente de la temperatura. Se mide con células de conductividad o con sensores conductimétricos.
Un parámetro muy informativo es la llamada conductividad equivalente de una disolución, que es proporcional al número de equivalentes por litro de la(s) especie(s) conductora(s) (un equivalente de un ion es su peso atómico dividido por su valencia iónica) e inversamente proporcional a la conductividad. Cuando este parámetro se extrapola a dilución infinita (es decir, a concentración nula) se obtiene la conductividad equivalente a dilución infinita, un parámetro que es específico de cada especie conductora y además es aditivo; es decir, la conductividad equivalente a dilución infinita de una disolución es la suma de las conductividades equivalentes a dilución infinita de cada uno de sus iones.
Una de las aplicaciones más útiles de la medida de la conductividad es la valoración conductimétrica. Consiste en detectar el punto de equivalencia en una valoración por un cambio brusco de la conductividad una vez que dicho punto se ha superado.
La conductimetría tiene importantes aplicaciones en medio ambiente. La conductividad del agua es una medida objetiva de su pureza. También ofrece un método rápido aunque aproximado de evaluar la cantidad de sólidos disueltos totales. Mediante sensores conductimétricos se pueden determinar gases. Por otro lado, la medida de la conductividad de terrenos es un método utilísimo para detectar materiales metálicos y ciertos vertidos enterrados.
MATERIAL DIDÁCTICO
Ejemplos de aplicación de las técnicas de corriente eléctrica a problemas ambientales:
^^^Volver al índice
Tema 11 (TÉCNICAS RADIOQUÍMICAS Y TÉRMICAS Y MÉTODOS
AUTOMÁTICOS)
-
COMENTARIOS
Técnicas radioquímicas
Como se sabe, la radiactividad es un problema ambiental de primer orden. No tanto la natural, que «siempre ha estado ahí», sino la inducida por las actividades humanas. Por eso, la detección de sustancias radiactivas en el medio ambiente es un reto fundamental en química analítica.
Los núcleos radiactivos, al desintegrarse, emiten distintos tipos de radiaciones y partículas: negatrones, positrones –ambas se engloban bajo el nombre de partículas beta–, partículas alfa y rayos gamma. También pueden capturar electrones e incluso romperse (fisionarse). Cada núcleo (13C, 18O, 15N…) se desintegra de un modo específico que es independiente de su estado físico o químico (por ejemplo, un núcleo de C radiactivo se desintegra del mismo modo cuando forma parte de una molécula de CO2 que cuando está contenido en una molécula de benceno). Este es el fundamento de las técnicas espectroscópicas basadas en la radiactividad.
Los tres tipos de espectros más útiles son el de rayos gamma, el de partículas alfa y el de negatrones, por ese orden. En el espectro de rayos gamma (formalmente parecido a un espectro atómico) se observan picos cuyos valores de energía e intensidad son específicos de cada núcleo. La intensidad se mide en cuentas por segundo (es decir, número de fotones que el detector cuenta cada segundo) y la energía es del orden de 100 o 1000 keV. Su interpretación es sencilla. Como también lo es la de los espectros de partículas alfa, aunque en este caso se producen bandas (no muy anchas) correspondientes a energías algo mayores. Finalmente, los espectros de negatrones consisten en bandas extraordinariamente anchas. Estos son, por lo tanto, menos útiles.
La radiactividad se mide con detectores de ionización de gas (para detección rutinaria de partículas alfa y beta), de centelleo (para partículas beta y rayos gamma) o de semiconductores (rayos gamma). El conocido detector de Geiger-Müller es de ionización de gas. Para cuantificar las muestras, estas se disponen en capas delgadas para evitar el fenómeno de la autoabsorción. Las áreas de los picos son proporcionales a la concentración.
Lo comentado hasta aquí se refiere a la medida de la radiactividad de las muestras. Pero las técnicas radioquímicas también se emplean para estudiar muestras no radiactivas. Así, se puede bombardear con neutrones o rayos gamma una muestra para hacerla radiactiva y posteriormente medir la radiactividad inducida, que es proporcional a la cantidad del analito en la muestra. Esta técnica se llama activación neutrónica (o fotónica, si se hace con rayos gamma). Permite determinar hasta 70 elementos químicos con buenos límites de detección.
Otra técnica radioquímica aplicable a muestras no radiactivas es la dilución isotópica. Se trata de añadir a la muestra una cantidad conocida de isótopo radiactivo del analito. Siguiendo ciertos procedimientos químicos y midiendo la radiactividad de distintas fracciones se puede conocer cuánto analito había en la muestra aplicando una sencilla fórmula.
Las aplicaciones ambientales son obvias: medir la radiactividad natural o antropogénica. Para ello incluso se han diseñado sistemas aproximados pero baratos como tiras de policarbonato (que sufren daños físicos microscópicos si hay radiactividad) o ciertas películas impresionables. Pero también se pueden determinar, por activación neutrónica o dilución isotópica, gran cantidad de muestras no radiactivas de suelos, aguas y atmósfera. Otras aplicaciones son el estudio del subsuelo (densidad, porosidad, humedad…) por irradiación con fotones gamma o bombardeo con neutrones.
Técnicas térmicas
Existen unas técnicas que permiten obtener información de las muestras estudiando cómo responden al calentamiento o enfriamiento; se llaman por ello térmicas.
La termogravimetría estudia cómo cambia la masa de un compuesto químico al elevar su temperatura. El cambio de masa se debe a reacciones químicas o cambios físicos que experimenta el compuesto. Por ejemplo, puede perder agua de cristalización, agua de hidratación, CO2, NH3…; se puede descomponer, oxidar; se le pueden desorber otras sustancias, etc.
La muestra se introduce en el termogravímetro, que es un instrumento que cuenta con un horno, una balanza analítica y, eventualmente, un sistema dispensador de gases (oxígeno, gases inertes…). Se obtiene así el termogravigrama, que es una representación gráfica de la masa de la muestra frente a la temperatura. Normalmente se obtiene una curva que presenta saltos, indicando cada uno un proceso. La derivada de la curva correspondiente es el termogravigrama diferencial, compuesto por una serie de bandas, cada una de las cuales revela el proceso fisicoquímico correspondiente. Cada especie química da su propio termogravigrama, por lo que este es útil para identificarla. Usando patrones, también se puede cuantificar. En medio ambiente esta técnica es útil para determinar el contenido de agua, materia orgánica y carbonatos en arcillas y, en general, en suelos.
El análisis térmico diferencial consiste en medir la diferencia de temperatura entre una muestra y un material de referencia cuando se someten ambos al mismo programa de calentamiento. Representando la diferencia de temperatura entre muestra y referencia frente a la temperatura de la referencia se pueden detectar procesos fisicoquímicos en la muestra como fusión, vaporización, sublimación, absorción, desorción, deshidratación, oxidación, reducción, ya que algunos de estos procesos son exotérmicos (generan calor) y otros endotérmicos (lo absorben). En el momento en que se produce un proceso exotérmico la muestra se calienta más que la referencia por el propio calor que genera, y al contrario cuando el proceso es endotérmico. Estos procesos, exo y endotérmicos, producen curvas con bandas o picos de distinto signo en el registro.
El registro es, lógicamente, específico de cada compuesto químico, lo que permite en muchos casos identificarlo y también cuantificarlo si se usan patrones. No obstante, hay que tener presente que la técnica no es muy repetible por la dificultad de conseguir que la muestra tenga la misma temperatura en todas sus partes. Para lograrlo, hay que tomar muy pequeñas cantidades de muestra bien homogeneizada.
La técnica se lleva a cabo en el analizador térmico diferencial, formado por un horno que contiene dos platillos, para la muestra y la referencia, y sendos termopares que miden las temperaturas de ambas. Es especialmente útil para estudiar polímeros (plásticos). En medio ambiente se ha aplicado con éxito a muestras carbonosas, arcillas y suelos. En química teórica la técnica es útil para obtener diagramas de fases y hacer medidas cinéticas.
La calorimetría diferencial de barrido proporciona información semejante a la de la técnica anterior. Pero en este caso lo que se mide es la diferencia de calor que es preciso suministrar a una muestra respecto a una referencia para conseguir que ambas mantengan la misma temperatura siguiendo un programa de calentamiento. Cuando la muestra experimenta un proceso exotérmico, es decir, genera calor por sí misma, el aparato disminuye el flujo hacia ella (manteniendo constante el de la referencia) para que su temperatura no suba más que la de la referencia. Cuando la muestra experimenta un proceso endotérmico el sistema opera al revés. Esta técnica prácticamente tiene las mismas aplicaciones cualitativas y cuantitativas que la anterior, pero se emplea más, entre otras razones porque ofrece límites de detección más bajos. En medio ambiente se ha aplicado, por ejemplo, para diferenciar fracciones orgánicas del suelo.
Otras técnicas térmicas son la valoración termométrica y la entalpimetría de inyección directa. La primera permite detectar el punto de equivalencia de una valoración midiendo el cambio de temperatura conforme se añade valorante. La segunda mide el cambio de temperatura de una muestra tras añadirle un reactivo; el cambio es proporcional a la concentración de uno o varios analitos. Por valoración termométrica se puede diferenciar la dureza de agua debida a calcio de la debida a magnesio. Por entalpimetría se puede conocer el contenido de SO2 y CO2 en muestras de aire.
Por otro lado, la simple medida de la temperatura del subsuelo da información ambiental. Por ejemplo, se pueden determinar las recargas estacionales de los acuíferos.
Métodos automáticos
Los laboratorios ambientales a menudo tienen que analizar gran cantidad de muestras por procedimientos de rutina. Para hacerlo más rápidamente y de modo más repetible son útiles los sistemas automáticos. Los hay de dos tipos generales: discontinuos y continuos. Los primeros son como robots. Normalmente pueden tomar muestras automáticamente de una bandeja, prepararlas, tomar alícuotas, disolverlas, separar los analitos de la matriz, medir una señal analítica determinada, calibrar con patrones y presentar los datos. En medio ambiente se han ideado autoanalizadores discontinuos para medir el contenido de C, H, N, O y S de una muestra; o el de C y N disueltos. También pueden medir el pH y parámetros significativos como el C total, C orgánico, C inorgánico, N total, etc.
En los analizadores discontinuos cada muestra se encuentra en su propio recipiente. Sin embargo, en los continuos todas las muestras se inyectan secuencialmente en un flujo de un líquido portador. En la variedad de flujo segmentado, cada inyección se separa de la siguiente mediante una burbuja de aire que evita la mezcla de las muestras. En la de inyección en flujo continuo no se hace esta separación pero se procura que la hidrodinámica del flujo sea tal que impida la dispersión de cada muestra en el flujo y la consiguiente mezcla con la precedente y la antecedente. Algunos equipos están dotados con módulos especiales (dializadores, reductores, extractores, destiladores…) según los tratamientos que cada muestra precise.
En general, en ambos casos las muestras se mezclan en un serpentín con reactivo(s) para generar un producto que se analiza por el procedimiento químico apropiado. A menudo, lo que se persigue es que se forme un producto coloreado que se pueda cuantificar por absorción UV-visible. En otros casos se usan detectores de masas, de emisión atómica, electroquímicos o basados en cualquier otra técnica instrumental. En la inyección en flujo no se suele esperar a que la reacción se complete; basta hacer el análisis siempre dejando transcurrir el mismo tiempo desde que el reactivo se mezcla con la muestra. Otras veces el flujo se detiene periódicamente para aumentar el tiempo de mezcla de la muestra con su reactivo (flujo detenido). La técnica es ideal para realizar valoraciones automáticas.
Las aplicaciones ambientales de las técnicas automáticas de flujo continuo son numerosísimas.
Por otra parte, relacionado con el concepto del flujo continuo está el de los métodos cinéticos de análisis. Se emplean para determinar analitos que forman productos con algunos reactivos pero que lo hacen muy lentamente. El fundamento de estos métodos es medir la velocidad de reacción en los primeros momentos de esta. Este parámetro (la velocidad inicial) es específico de cada reacción y también depende de las concentraciones, por lo que tiene propiedades identificativas y cuantificadoras. Se puede trabajar tanto con flujo continuo como detenido.
Guardan cierta analogía con los métodos automáticos los procedimientos de análisis mediante tiras reactivas. Estas, impregnadas de reactivos, dan diferentes colores según el analito que está presente. Aunque los resultados son semicuantitativos, en medio ambiente son de gran utilidad, al menos prospectiva.
MATERIAL DIDÁCTICO
Ejemplos de aplicación de la potenciometría a problemas ambientales:
^^^Volver al índice
Tema 12 (TÉCNICAS CROMATOGRÁFICAS)
-
COMENTARIOS
Bases de la cromatografía
La mayor dificultad con que el analista se encuentra cuando se ha de estudiar muestras ambientales suele ser su tremenda complejidad. Aunque existen tratamientos químicos que pueden aislar los analitos de interés, lo mejor es llevar a cabo un tratamiento fisicoquímico: la cromatografía. Hay muchas y variadas técnicas cromatográficas, pero el objetivo de todas es separar las sustancias que forman una mezcla y enviarlas secuencialmente a un detector para que las determine y cuantifique. En general, estas técnicas se pueden clasificar en varias familias: cromatografía de gases, de líquidos, mediante fluidos supercríticos y en capa fina.
Todas se basan en el mismo fenómeno: permitir que las sustancias que forman una mezcla entren en contacto con dos fases (un líquido y un gas, un sólido y un líquido, etc.). Una de las fases es estática (no se mueve) y tenderá a retener las sustancias en mayor o menor grado; la otra, móvil, tenderá a arrastrarlas. Cada sustancia química tiene distinta tendencia a ser retenida y a ser arrastrada. Dicho más correctamente, cada sustancia tiene distinto coeficiente de distribución entre las dos fases. El coeficiente de distribución es una medida de la tendencia relativa a quedar en una fase u otra.
Se opera de modo que en una primera etapa se deja que las sustancias que forman la mezcla entren en contacto con la fase estática. Cada sustancia de la mezcla tendrá una mayor o menor afinidad por esta fase. Después se hace pasar la otra fase, que arrastrará en mayor grado las sustancias menos afines por la primera. Típicamente, el proceso se lleva a cabo en una columna. Dentro de ella está fijada la fase estática y a través de ella se hace pasar la fase móvil, que se llama eluyente.
La muestra se introduce por la cabeza de la columna. Después se hace pasar el eluyente. Por el otro extremo empezarán a salir los distintos componentes de la mezcla, que podrán ser detectados con el detector correspondiente (de conductividad, ultravioleta, de masas…; en realidad, cualquier técnica de las estudiadas hasta aquí puede servir de base de detección, y también técnicas específicas ideadas especialmente para cromatografía). Existen dos tipos de detectores: universales y selectivos; los primeros detectan casi cualquier sustancia (aunque en distinto grado).
Un registro gráfico en el que aparecen una serie de picos (uno por cada especie separada) frente al tiempo de duración del experimento constituye un cromatograma. Las sustancias que dan lugar a cada pico pueden reconocerse por dos métodos. Uno es el análisis directo por alguna técnicas muy rápidas (IR, masas…); se realiza un espectro de la sustancia que eluye en cada momento y así se determina su naturaleza. Otro es usar patrones que se hacen pasar por la columna para medir el tiempo que tardan en ser eluidos. De este modo, comparando tiempos en los patrones y en la muestra se pueden identificar los componentes de la muestra, ya que el tiempo que tardan en eluir es una característica específica de cada sustancia. La cuantificación se lleva a cabo midiendo las áreas de los picos y comparándolas con las de patrones. Lo mejor es emplear el método del patrón interno para soslayar los inconvenientes de baja repetibilidad propios de las técnicas cromatográficas.
Ahora bien, este tiempo depende de muchas condiciones: el flujo (velocidad) de la fase móvil, su composición (la naturaleza del diluyente, que puede ser puro o una mezcla de varios), la temperatura (puede mantenerse constante o programarse para que varíe controladamente a lo largo del experimento), el tamaño de las partículas de la fase fija, la cantidad de muestra, etc. Cada experimento requiere unas condiciones diferentes. Si se observa que los picos solapan, deben cambiarse las condiciones hasta conseguir una buena eficacia separadora.
Cromatografía de gases
En cromatografía de gases la fase móvil es un gas llamado portador. La otra suele ser un líquido adsorbido sobre un sólido (cromatografía de gases gas-líquido) o, bastante menos comúnmente, un sólido (cromatografía de gases gas-sólido). Las columnas son finas y muy largas (por eso se arrollan). En esta técnica el control de la temperatura es fundamental, por lo cual la columna debe estar bien termostatizada (dentro de una estufa) durante todo el proceso. Existen dos tipos de columnas: rellenas (de fase estática) y abiertas (la fase estática está adherida a las paredes). Estas últimas son capilares. Su eficacia es mejor.
Como detectores se suele emplear el de conductividad térmica (que es universal y mide lo que baja la conductividad térmica del gas portador al mezclarse con cada especie separada); el de ionización en llama (la especie separada se quema, lo que en ciertos casos genera cargas eléctricas y una pequeña corriente proporcional a la concentración); o el de captura electrónica (basado en técnicas radiactivas). También se usan detectores generales como el de masas, IR, emisión atómica o algunos muy específicos como el de quimioluminiscencia de azufre.
La técnica ofrece unos excelentes resultados cuando se acopla con un espectrómetro de masas porque cada sustancia que va eluyendo puede ser fácilmente identificada. También se obtiene mucha información cuando se acopla al cromatógrafo un espectrómetro IR o uno de RMN.
La cromatografía de gases se aplica sobre todo a muestras orgánicas volátiles o volatilizables por derivatización. Pueden estar en estado sólido, líquido o, por supuesto, gas, pero muestras líquidas y sólidas deben vaporizarse previamente. La modalidad de gas-sólido permite detectar y cuantificar gases atmosféricos, por ejemplo.
Cromatografía de líquidos
En cromatografía de líquidos la fase móvil es líquida. Las columnas son mucho más cortas que en gases. El control de la temperatura no es tan crítico, pero sí ha de serlo el de la presión. Se ejercen presiones muy altas para hacer pasar la fase móvil (un líquido) a través de la estática (un sólido). Se aplica a especies no volátiles o térmicamente inestables.
El instrumento consta, además de la columna y detector, de una bomba de alta presión, un sistema desgasificador –los gases ensanchan las bandas– y una columna de sacrificio (precolumna) para eliminar materia en suspensión en los disolventes, polvo, etc., y un programador de disolventes para mezclarlos según se precise y de este modo mejorar el cromatograma.
El detector que más se emplea en cromatografía de líquidos es el de absorción UV. También pueden usarse un detector de IR, de masas, de RMN, de fluorescencia, de índice de refracción (este es muy universal) y en ciertos casos de conductividad iónica.
Según la propiedad fisicoquímica que se aprovecha para separar las especies de una mezcla se distinguen cuatro tipos básicos de cromatografía de líquidos: de reparto, de adsorción, de intercambio iónico y de exclusión por tamaño.
En la de reparto, la fase estática es un líquido unido a un sólido. Si este líquido es más polar que el de la fase móvil, la modalidad se llama de fase normal; si es no polar y la fase móvil es polar, se llama de fases invertidas. Esta se aplica más. En la de adsorción la fase fija es sólida; por ello la técnica se denomina también cromatografía líquido-sólido. La de intercambio iónico se emplea sobre todo para separar especies iónicas. La fase es una resina de intercambio iónico o material análogo. En la de exclusión por tamaño la fase fija es un material poroso; su función es retener en sus microtubos las especies de bajo peso molecular y dejar pasar moléculas más grandes. De este modo, las mayores eluyen antes.
Cromatografía mediante fluidos supercríticos
En este técnica se emplea como fase móvil una sustancia en condiciones de temperatura y presión tales que sea supercrítica (es decir, tenga propiedades intermedias entre un gas y un líquido), como el CO2 a más de 31 ºC y presión superior a 73 atm. Esta técnica reúne algunas ventajas de la cromatografía de gases y de la de líquidos. Es útil para compuestos no volátiles o térmicamente inestables que no se detectan bien en cromatografía de líquidos. Lógicamente, en esta técnica es fundamental tanto el control de la presión como el de la temperatura. Una gran ventaja es que cuando el eluido se pone a presión y temperatura ambiente, el eluyente se evapora. Como detectores se suelen usar tanto los de cromatografía de gases como los de líquida.
Cromatografía en capa fina
En vez de en columna, se puede llevar a cabo la separación cromatográfica en un plano (un papel o una placa especial). La muestra se aplica sobre la fase móvil y sus componente son arrastrados hacia arriba, por capilaridad, por la fase móvil (líquida), o bien hacia abajo por gravedad o impulsados por una diferencia de potencial eléctrico. Se deja que los componentes de la mezcla se muevan a lo largo de la placa (unos más y otros menos, según su factor de retención o factor de retención relativo (frente a un patrón). Para medir este parámetro es necesario ver hasta dónde llega la mancha de cada analito. Para ello, la placa se ha de «revelar» mediante alguna sustancia química que forme algún productos coloreado o fluorescente con los analitos.
La identificación de los analitos de cada mancha también puede hacerse raspándola y analizándola por cualquier técnica. La cuantificación suele hacerse con ayuda de un densitómetro, un aparato que mide la densidad óptica o intensidad de cada mancha, que es proporcional a la concentración del analito que la genera.
Existen variedades de la técnica de capa fina como la de alta resolución, la bidimensional y la de fases invertidas.
Electrocromatografía capilar y electrofresis capilar
Otra técnica cromatográfica consiste en introducir la muestra en un capilar relleno y someterla a una alta diferencia de potencial. Eso crea un flujo electroosmótico que arrastra el disolvente desde el ánodo hacia cátodo. Cerca del cátodo se coloca un detector que va analizando los analitos que pasan por el capilar. La técnica, que se llama electrocromatografía capilar, Se puede aplicar a especies iónicas o no porque todas son arrastradas.
Una técnica relacionada, que no es cromatográfica porque no se basa en la diferencia de afinidad por dos fases, es la electroforesis capilar. El dispositivo es análogo al de técnica anterior, pero el capilar se rellena simplemente con un tampón, o bien se realiza en una placa. El fundamento de la técnica es que las especies avanzan impulsadas por el flujo electroosmótico pero también por atracción electrostática (los cationes se aceleran hacia el cátodo pero los aniones se retrasan por la atracción del ánodo). La técnica es útil para separar especies iónicas. Se pueden detectar por espectroscopía UV-visible, fluorescencia, técnicas radioquímicas o electroquímicas, espectrometría de masas o simple tensión.
En medio ambiente
La cromatografía se ha aplicado extensivamente en medio ambiente: gases de invernadero, compuestos orgánicos volátiles, hidrocarburos en suelos y agua, bifenilos policlorados, halofenoles, metales… Los cromatógrafos de gases acoplados con espectrómetros de masas se pueden «miniaturizar» y transportar al campo. También se han ideado práctico sensores cromatográficos.
MATERIAL DIDÁCTICO
Ejemplos de aplicación de las técnicas cromatográficas a problemas ambientales:
^^^Volver al índice
Comentarios desactivados en Explicaciones, resúmenes de los temas y material didáctico adicional
Tema 1
- Página 552. La solución al ejercicio 1.2 no debe considerarse la b, sino la d. Además, es preferible sustituir la explicación de la resolución del ejercicio que se da por esta otra:
d. En general, cuando la fuente de contaminación está muy localizada en un punto y se puede extender en todas direcciones, el muestreo recomendable por su mejor relación resultados/coste es el polar. El inconveniente principal (que es que no todos los puntos dibujados sistemáticamente o seleccionados aleatoriamente en un plano de una ciudad son fácilmente muestreables, ya que algunos pueden coincidir con tejados o zonas particulares) no impide hacer un muestreo de este tipo, pues los puntos de muestreo no tienen por qué ser exactamente los inicialmente elegidos (no se espera un error apreciable por muestrear unos metros más allá del punto marcado en el plano). Un muestreo aleatorio nunca se puede decir que no dé buenos resultados, pero en este caso no es recomendable por la dificultad anterior y por su costo (se necesita tomar muchas muestras). El muestreo a juicio sería plausible si la ciudad estuviera sometida continuamente a vientos de una determinada componente, de modo que pudiéramos suponer que el contaminante solo se transporta en una dirección determinada.
Tema 3
- Página 155. Al inicio del apartado 3.6.2, donde dice: Sea P0 la potencia de la radiación de determinada l que sale de una fuente e incide en una muestra, y P la que sale de la muestra y alcanza el detector, de la misma l. El cociente P/P0 es la fracción de potencia total absorbida y se llama transmitancia (T)debe decir:Sea P0 la potencia de la radiación de determinada l que sale de una fuente e incide en una muestra, y P la que sale de la muestra y alcanza el detector, de la misma l. El cociente P/P0 es la fracción de potencia total transmitida y se llama transmitancia (T)
- En esa misma página, arriba, al final del párrafo introductorio del apartado 3.6, donde dice:en absorción, la relación lineal lo es entre la concentración y el logaritmo de la fracción de potencia radiante absorbida por la muestra.debe decir:en absorción, la relación lineal lo es entre la concentración y el logaritmo de la fracción de potencia radiante transmitida por la muestra.
Tema 4
- Página 187. En el último renglón se lee «radiación de sincrotón». Es «sincrotrón«.
Tema 9
- Página 411. En el antepenúltimo párrafo se lee: «El electrodo selectivo de protones se conoce por el nombre de electrodo de vidrio porque se basa en una membrana de un material vítreo. Merece una atención especial». Aunque no se trata de una errata, la frase estaría mejor escrita así: «Se emplea para ello el llamado electrodo de vidrio porque se basa en una membrana de un material vítreo. (Existen electrodos de vidrio para determinar otros cationes, como Na+ o Ag+)».
- Página 413. La ecuación 9.18 no es pH = 0,059(K – Epila(medido)), sino pH = (K – Epila(medido))/0,059.
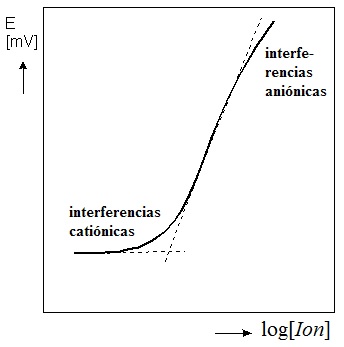 Página 415. En el eje de abscisas de la gráfica 9.12 debe decir -pIon, no pIon o, lo que sería aun más claro: log[Ion]. Lo mismo en el pie de la figura.
Página 415. En el eje de abscisas de la gráfica 9.12 debe decir -pIon, no pIon o, lo que sería aun más claro: log[Ion]. Lo mismo en el pie de la figura.- Página 422. La pregunta 9.5 («Uno de los siguientes dispositivos no es adecuado para medir directamente fluoruros en agua.») debe sustituirse por esta otra: «Solo uno de los siguientes dispositivos es adecuado para medir directamente cloruros en agua». La respuesta correcta (página 567) sería la b y debería estar acompañada por este texto: «b. El electrodo de vidrio mide la concentración de protones y otros cationes. Los electrodos señalados en a y c son específicos para el fluoruro».

Tema 12
- Página 522. En el segundo renglón, donde dice «cada muestra» debería decir «cada analito».
- Página 534. En el penúltimo párrafo, donde dice «un disolvente orgánico polar con metanol» debe decir «un disolvente orgánico polar, como el metanol,».
- Página 545. En el antepenúltimo párrafo, donde dice «separar y cuantificar los aniones y cationes más comunes (halogenuros, NO3–, NO2–, SO42- o PO43…» falta la carga negativa del grupo fosfato (PO43-).
Glosario
- Página 578. En la definición de isótopo, donde se dice «se conocen dos isótopos estables del cloro: 35Cl y 37Cl. El primero tiene 35 neutrones y el segundo 37″ debería decirse «El primero tiene 18 neutrones y el segundo 20».
Tema 1
- La solución al ejercicio 1.2 no debe considerarse la b, sino la d. Además, es preferible sustituir la explicación de la resolución del ejercicio que se da por esta otra:
«d. En general, cuando la fuente de contaminación está muy localizada en un punto y se puede extender en todas direcciones, el muestreo recomendable por su mejor relación resultados/coste es el polar. El inconveniente principal (que es que no todos los puntos dibujados sistemáticamente o seleccionados aleatoriamente en un plano de una ciudad son fácilmente muestreables, ya que algunos pueden coincidir con tejados o zonas particulares) no impide hacer un muestreo de este tipo, pues los puntos de muestreo no tienen por qué ser exactamente los inicialmente elegidos (no se espera un error apreciable por muestrear unos metros más allá del punto marcado en el plano). Un muestreo aleatorio nunca se puede decir que no dé buenos resultados, pero en este caso no es recomendable por la dificultad anterior y por su costo (se necesita tomar muchas muestras). El muestreo a juicio sería plausible si la ciudad estuviera sometida continuamente a vientos de una determinada componente, de modo que pudiéramos suponer que el contaminante solo se transporta hacia una dirección determinada».
Tema 2
- En la página 111, al final, se dice: «…si c0 en el patrón original es 0.1 M, las concentraciones de analito procedente del patrón en cada uno los recipientes (de izquierda a derecha en la figura 2.11) serán 0.01, 0.02, 0.03, 0.04 y 0.05 M». Lo correcto es: 0.00, 0.01, 0.02, 0.03 y 0.04 M.
- En las páginas 123-124 el ejercicio 2.6 debe renumerarse como 2.7 y viceversa.
Tema 3
- En el segundo párrafo de la página 147, donde dice «figura 3.21-derecha» debe decir «figura 3.22-derecha».
- Página 155. Al inicio del apartado 3.6.2, donde dice:
Sea P0 la potencia de la radiación de determinada l que sale de una fuente e incide en una muestra, y P la que sale de la muestra y alcanza el detector, de la misma l. El cociente P/P0 es la fracción de potencia total absorbida y se llama transmitancia (T), debe decir: «Sea P0 la potencia de la radiación de determinada l que sale de una fuente e incide en una muestra, y P la que sale de la muestra y alcanza el detector, de la misma l. El cociente P/P0 es la fracción de potencia total transmitida y se llama transmitancia (T)». - En esa misma página, arriba, al final del párrafo introductorio del apartado 3.6, donde dice:
«en absorción, la relación lineal lo es entre la concentración y el logaritmo de la fracción de potencia radiante absorbida por la muestra», debe decir: «en absorción, la relación lineal lo es entre la concentración y el logaritmo de la fracción de potencia radiante transmitida por la muestra«. - En la página 159, al principio del apartado 3.6.3 se dice: Aunque las expresiones [3.4] y [3.6] predicen teóricamente relaciones lineales entre la potencia absorbida o la absorbancia y la concentración…
pero debe decir: «Aunque las expresiones [3.4] y [3.6] predicen teóricamente relaciones lineales entre la potencia emitida o la absorbancia y la concentración…». - En la resolución del ejercicio 3.7 se ha empleado el valor de temperatura de 10000 K en vez del de 8150 que aparece en el enunciado.
Tema 4
- Página 189. En el penúltimo renglón se lee «radiación de sincrotón». Es «sincrotrón«.
Tema 5
- En la página 235 en la segunda línea, pone «figura 5.9». Lo correcto es: «figura 5.10».
Tema 7
- En la página 344, en su penúltima línea, dice «10, 4, 4, 4 y 3». Lo correcto es: «10, 4, 4, 4 y 6».
Tema 8
- En la página 372, en la línea 14ª, dice «dióxido de tritio», pero debe decir «óxido de tritio».
Tema 9
- En la página 407, en la figura, en la reacción del Zn, en la parte de la derecha de la flecha, el 2 que aparece como subíndice del Zn debe aparecer como superíndice.
- En la página 414, inmediatamente antes del epígrafe 9.3.1.2 dice: «De las reacciones que aparecen en la tabla se dan más fácilmente que la reducción del hidrógeno todas las reacciones de reducción que figuran por encima de la de él; es decir, todas las que tienen un valor E0 < 0″. Pero en realidad debe decir: «De las reacciones que aparecen en la tabla se dan más fácilmente que la reducción del hidrógeno todas las reacciones de reducción que figuran por debajo de la de él; es decir, todas las que tienen un valor E0 > 0″.
- Página 421. En el antepenúltimo párrafo se lee: «El electrodo selectivo de protones se conoce por el nombre de electrodo de vidrio porque se basa en una membrana de un material vítreo. Merece una atención especial». Aunque no se trata de una errata, la frase estaría mejor escrita así: «Se emplea para ello el llamado electrodo de vidrio porque se basa en una membrana de un material vítreo. (Existen electrodos de vidrio para determinar otros cationes, como Na+ o Ag+)».
- Página 425. En el eje de abscisas de la gráfica 9.12 debe decir -pIon, no pIon o, lo que sería aun más claro: log[Ion]. Lo mismo en el pie de la figura.
- Página 433. La pregunta 9.5 («Uno de los siguientes dispositivos no es adecuado para medir directamente fluoruros en agua.») debe sustituirse por esta otra: «Solo uno de los siguientes dispositivos es adecuado para medir directamente cloruros en agua». La respuesta correcta (página 581) sería la b y debería estar acompañada por este texto: «b. El electrodo de vidrio mide la concentración de protones y otros cationes. Los electrodos señalados en a y c son específicos para el fluoruro».
Tema 10
- En el penúltimo párrafo de la página 464, donde dice «Esto es así porque Λ depende de la velocidad de migración de los iones y esta es mayor a menor dilución porque los iones encuentran menos impedimento en su recorrido» debe decir «Esto es así porque Λ depende de la velocidad de migración de los iones y esta es mayor a mayor dilución porque los iones encuentran menos impedimento en su recorrido».
- En el tercer párrafo de la página 467, donde dice «Basta medir la conductividad G«, debe decir «Basta medir la conductancia G«. En ese mismo párrafo, donde dice «conductancia equivalente» debe decir «conductividad equivalente».
- En la página 470, en el renglón 12, donde dice polarimetría debe decir polarografía.
Tema 11
- En la página 498, casi al comienzo, donde dice «…la representación de la masa frente al tiempo constituye un termogravigrama» debe decir más correctamente «…la representación de la masa frente a la temperatura constituye un termogravigrama» (como se puede observar en la figura que aparece más abajo).
- En la página 501, en la 2ª línea tras la figura, dice «todos exotérmicos, todos exotérmicos o de uno y…». Lo que debe decir es: «todos exotérmicos, todos endotérmicos o de uno y…».
Tema 12
- Página 423. La ecuación 9.18 no es pH = 0,059(K – Epila(medido)), sino pH = (K – Epila(medido))/0,059.
- Página 425. En el eje de abscisas de la gráfica 9.12 debe decir -pIon, no pIon o, lo que sería aun más claro: log[Ion]. Lo mismo en el pie de la figura. (Ver imagen a la derecha).
- Página 433. La respuesta b a la pregunta 9.5 («Electrodo de segunda clase de plata») debe sustituirse por esta otra: «Un electrodo selectivo de iones especialmente diseñado».
- En la página 534, en el segundo renglón, donde dice «cada muestra» sería más propio decir «cada analito».
- En la página 545 (punto 12.4.4) se dice que «…se pueden distinguir varios tipos generales de cromatografía líquida en columna y en capa fina: de reparto, de adsorción, de intercambio iónico y de exclusión por tamaño«. Hay que aclarar que esas son variedades solo de la cromatografía en columna, no de la capa fina.
- Página 546. En el penúltimo párrafo, donde dice «un disolvente orgánico polar con metanol» debe decir «un disolvente orgánico polar, como el metanol,».
- En la página 550, en el quinto renglón, donde dice «lápiz de plomo» debe decir «lápiz de grafito».
- Página 557. En el antepenúltimo párrafo, donde dice «separar y cuantificar los aniones y cationes más comunes (halogenuros, NO3–, NO2–, SO42- o PO43…» falta la carga negativa del grupo fosfato (PO43-).
Glosario
- Página 592. En la definición de isótopo, donde se dice «se conocen dos isótopos estables del cloro: 35Cl y 37Cl. El primero tiene 35 neutrones y el segundo 37″ debería decirse «El primero tiene 18 neutrones y el segundo 20».
Comentarios desactivados en Erratas de la primera edición del libro (2007)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}